The major regulatory element of the HPA axis is the glucocorticoid receptor, whose functioning is moderated by chaperone proteins forming a molecular heterocomplex. Proper folding of the heterocomplex is required for ligand binding and translocation to the cell nucleus, where the activated glucocorticoid receptor exhibits its effects. In this respect, the FK506 binding protein 51 (FKBP5), coded by the
FKBP5 gene, is relevant as it is a co-chaperone to the heat shock protein 90 (hsp90) and modulates glucocorticoid receptor functioning (
6,
7). FKBP5 acts as an inhibitor of the glucocorticoid-receptor-mediated glucocorticoid action. In addition, glucocorticoids induce FKBP5 expression as part of an intracellular ultrashort negative feedback loop for glucocorticoid receptor activity (
8).
Our group (
9) and others (
10) have demonstrated that single-nucleotide polymorphisms (SNPs) in the
FKBP5 gene, including rs1360780, rs3800373, rs9296158, and rs9470080, are associated with an altered induction of FKBP5 expression by cortisol, leading to differences in glucocorticoid receptor sensitivity. These differences in receptor sensitivity correspond to other findings from our group, showing that three variants in the
FKBP5 gene (rs4713916, rs1360780, and rs3800373) are associated with an incomplete normalization of stress-elicited cortisol secretion in healthy subjects after psychosocial stress (
11). This suggests a genotype-dependent risk of chronically elevated plasma cortisol levels in the context of chronic or recurrent stress, which also may increase the risk of stress-related disorders, such as depression. This is further supported by the association of the same gene variants with unipolar depression (
12–
14), bipolar disorder (
15), and suicidality (
16). However, other investigators have not found a genetic main effect of
FKBP5 SNPs on depression (
9,
17). These inconsistencies could result from a failure to consider moderating influences, such as interactions with environmental factors (
18).
There are four studies published to date (
10,
12,
19,
20) exploring gene-environment interactions with regard to
FKBP5. Two of these indicated that
FKBP5 SNPs interact with child abuse to influence risk of posttraumatic stress disorder (PTSD) (
10,
20). Another study showed that the same
FKBP5 SNPs moderate the influence of childhood trauma on suicide risk (
19). Only one of these studies examined depression as an outcome, and it indicated a genetic main effect of one
FKBP5 SNP on depression in males only but no interaction with retrospectively assessed childhood problems (
12).
The aim of the present investigation was to examine the interaction between adverse life events and variants in the FKBP5 gene in relation to predicting the first occurrence of a major depressive episode.
Method
Design and Sample
The subjects were 996 respondents who participated in the molecular-genetic part of the Early Developmental Stages of Psychopathology study. This is a prospective longitudinal community study comprising a baseline and three follow-up waves covering an overall period of about 10 years. The representative baseline sample was drawn randomly from the 1994 government registries of all residents in Munich and surrounding counties between the ages of 14 and 24 years. The first follow-up, approximately 20 months after baseline, was conducted for subjects who were age 14–17 years at baseline; the second and third follow-ups included all subjects and occurred at approximately 42 and 101 months after baseline, respectively. The rate of retention from baseline (N=3,021) to the third follow-up (N=2,210) was 73.2%, and there was no selective attrition due to age, gender, geographic distribution, major depressive episodes, or the investigated adverse event categories (
21,
22).
The molecular-genetic part of the study was initiated at the third follow-up. There were 996 respondents who were European Caucasians of German nationality, according to interviewer ratings and respondents' self-reports on the country of origin for themselves, their natural parents, and their grandparents. Five individuals were of ethnicities other than European Caucasian and were excluded. Sociodemographic characteristics are presented in section SA1a of Appendix SA1 of the data supplement accompanying the online version of this article.
Of the 996 participants, 112 reported a lifetime history of a major depressive episode before or at baseline and were excluded from the investigation, leaving 884 individuals in the analysis. This prospective approach was done to prevent confounding of adverse event reports by depression status and to ensure that the adverse events occurred before the first major depressive episode, which is a prerequisite for causal interpretation.
The Early Developmental Stages of Psychopathology study was approved by the Ethics Committee of the Bayerische Landesaerztekammer Munich and the Medical Faculty of the Technische Universitaet Dresden. After complete description of the study to the subjects, written and oral informed consent was obtained.
Diagnostic Assessment
At each wave participants were interviewed with the computer-assisted Munich version of the Composite International Diagnostic Interview (M-CIDI) (
23), which allows for the standardized assessment of symptoms, syndromes, and diagnoses of DSM-IV disorders and information about onset, duration, and severity. It is supplemented by a respondent's booklet including disorder-specific questionnaires and symptom lists. Test-retest reliability and validity have been reported elsewhere (
24–
26). Trained clinical interviewers carried out the interviews face to face, mostly in the respondents' homes. At baseline, lifetime and 12-month disorders were assessed. At each follow-up interview, the M-CIDI covered the time interval since the previous interview.
Major depressive episodes and other mental disorders used as covariates (anxiety disorders, substance use disorders) were assessed by using the M-CIDI algorithms according to DSM-IV (see section SA1b of Appendix SA1 in the online data supplement). The main outcome measure, an incident major depressive episode, was defined as meeting the DSM-IV criteria for a major depressive episode for the first time during follow-up.
Adverse Life Events
In the evaluation of lifetime separation events, “death of a parent” (identified in 40 of 884 respondents, 4.5%) and “divorce of parents” (N=194, 21.9%) were assessed at baseline by using items from the family history section of the M-CIDI. The category “any separation event” covers “death” and “divorce” in any parent (N=223, 25.2%).
Lifetime trauma exposure before baseline was assessed at the beginning of the M-CIDI section for PTSD. Respondents were presented with a list of seven categories of traumatic events (see section SA1c of online Appendix SA1). “Any trauma” comprises all traumatic events (N=118, 13.3%). “Any severe trauma” includes only qualified traumas, which were reported to have caused intense fear, a feeling of helplessness, or horror (DSM-IV criterion A2 for PTSD) (N=90, 10.2%).
The category “any adverse event” (N=298, 33.7%) indicated exposure to any separation or traumatic event. Lifetime events preceding baseline occurred predominantly during childhood and adolescence (75% of severe traumas happened by age 16).
DNA Extraction and SNP Genotyping
The participants were asked to provide blood samples (40 ml) either at the Max Planck Institute of Psychiatry or at the practice of their general practitioner. Participants who did not comply were additionally asked for saliva samples, which were collected in four 10-ml vials containing an alcohol-based commercial mouthwash. DNA was extracted from fresh blood (for 885 of the 996 total respondents in the molecular-genetic study) or saliva (N=111).
Referring to our previous
FKBP5 studies (
9,
11), we selected five polymorphisms for genotyping: the three potentially functional SNPs (rs3800373, rs1360780, rs4713916) and two further SNPs (rs9296158, rs9470080) previously identified as relevant in a gene-environment analysis of PTSD (
10). These SNPs represent the
FKBP5 gene region at an average pairwise r
2 value (representing linkage disequilibrium) of 0.85 (see supplemental Figure SF1 in the data supplement accompanying the online version of this article). Genotyping was performed on a Sequenom platform employing the iPlex technology (Sequenom, San Diego). Hardy-Weinberg equilibrium was tested with Stata software (
27). Online supplemental Table ST1 presents information on the genotyped SNPs, Hardy-Weinberg equilibrium, minor allele frequency, and call rates (proportions of unambiguous genotypes). The linkage disequilibrium structure of the SNPs was determined by Haploview 4.0 (
28) with the r
2 statistic. It showed that four of the SNPs belong to the same linkage disequilibrium block and one (rs4713916) is highly associated with this block (see supplemental Figure SF1). The linkage disequilibrium structure in our sample is a good match with the linkage disequilibrium structures reported by the HapMap Project and in other samples (
9,
11). Power calculations are provided in section SA1d of the online supplemental Appendix SA1.
Statistical Analyses
The analyses followed the genotypic model, comparing the three possible genotypes with each other, and produced odds ratios based on logistic regressions adjusted for age and gender.
To examine interactions between
FKBP5 polymorphisms and adversity on an incident major depressive episode, an additive statistical interaction model was used. This model determines interaction effects as a deviation from additivity (instead of multiplicativity), which has been recommended for testing biologic interaction (
29–
32). A superadditive risk difference interaction means that the combined effect of two factors is larger (or smaller) than the sum of their individual effects (
30,
33). The risk difference has been proposed as an appropriate measure in terms of the standard models of causality (counterfactual causality and the sufficient component cause model) (
29–
31). We used the LOGISTIC procedure in Stata (
27) to fit logistic regressions and the MFX procedure to compute risk differences from the results. In the latter procedure, a method proposed by Joffe and Greenland was applied (
33,
34).
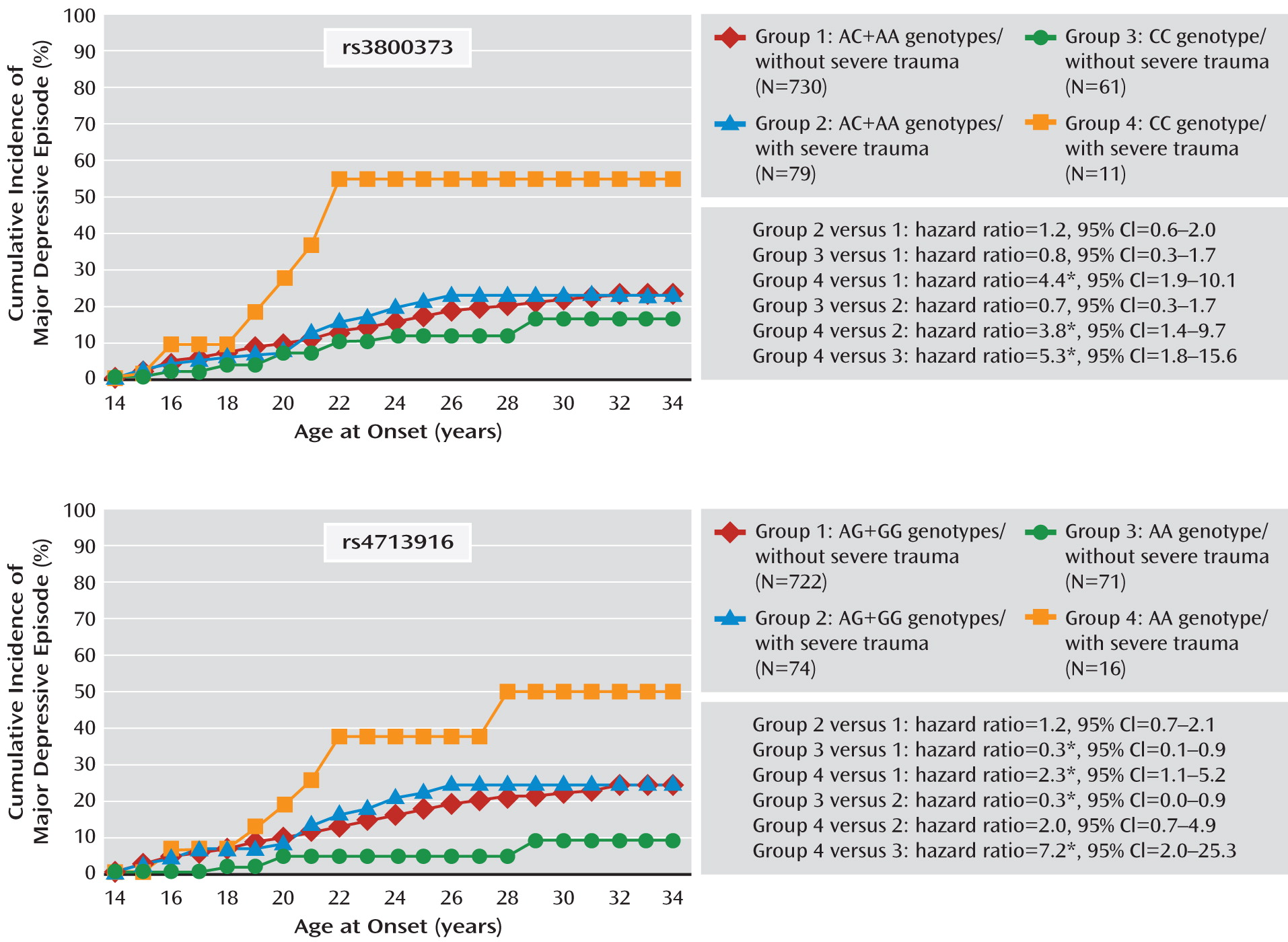
To illustrate the effect over the lifetime, age-specific cumulative lifetime incidences for major depressive episodes were calculated by means of the Kaplan-Meier method (
35, p. 7). Differences were tested with hazard ratios from stratified Cox regression (
35, p. 39).
We corrected for two independent event categories (separation and traumatic events) and for the five SNPs. Using the conservative Bonferroni correction for multiple testing, we set the level of significance to 0.005 (0.05/10).
To assure that interaction findings were not confounded by other preexisting disorders, we additionally controlled the logistic regressions for baseline anxiety and substance use disorders. Control variables were chosen a priori on the basis of previous research (
21,
36). All analyses were carried out with Stata 9.0 (
27).
Replication Studies
Samples.
Participants of the Dunedin Multidisciplinary Health and Development Study and the mothers of the participants in the Environmental Risk (E-Risk) Longitudinal Twin Study served as replication samples (
37). Briefly, the Dunedin sample was recruited from infants born in Dunedin, New Zealand, between April 1972 and March 1973 (N=1,037, 91% of eligible births; 52% male) and represents the full range of socioeconomic status in the general population of New Zealand's South Island. More than 90% of the participants are Caucasian. Assessments were conducted between the ages of 3 and 32, when 96% of the 1,015 study members still alive in 2004–2005 were interviewed. Evidence of childhood maltreatment was assessed between ages 3 and 11 years from five sources (shown in section SA1e of online supplemental Appendix SA1).
The base sample of the E-Risk study was formed in 1999–2000, when 1,116 mothers of 5-year-old twins (93% of eligible families) participated in home visit assessments. The participants represent the full range of socioeconomic status in the general population of England, and more than 90% are white. Four assessments were performed when the women were on average between 33 and 40 years (range=26–55 years, SD=5.8). The mothers completed the Childhood Trauma Questionnaire (
38) at an average age of 40 years (see section SA1e of online supplemental Appendix SA1).
In both replication samples, a major depressive episode during adulthood was the measured outcome and was evaluated with the Diagnostic Interview Schedule (
39) (see section SA1e of supplemental Appendix SA1).
DNA extraction and SNP genotyping.
In both replication samples, genotyping was restricted to the rs1360780 SNP because of technical and financial restrictions. We used the Applied Biosystems 7900HT TaqMan genotyping platform (Applied Biosystems, Foster City, Calif.). The genotypes were in Hardy-Weinberg equilibrium in both samples (Dunedin: p=0.94; E-Risk: p=0.88).
Statistical analyses.
For replicating the findings on gene-environment interactions, we performed a general log-linear model, which is a general procedure for model fitting and hypothesis testing in multidimensional contingency tables. We generated a three-dimensional contingency table that included the following dimensions: 1) lifetime major depressive episode: yes, no; 2) childhood maltreatment or trauma: “no, possible” or “some, severe”; and 3) genotype for rs1360780: CC, CT, TT. Hierarchical and nonstandard log-linear models were conducted by using the GENLOG procedure in SPSS 17.01 (IBM, Armonk, N.Y.). For the nonstandard log-linear analysis, the main effects of a lifetime major depressive episode, maltreatment, and genotype and the two-way interactions were defined as the base model. In the prediction model, the gene-environment interaction vector was additionally included to define the concurrent effects of the risk genotype (TT) and severe maltreatment on lifetime occurrence of a major depressive episode. Improvement in model fit was tested by using an incremental likelihood ratio test, and the significance of the gene-environment interaction vector was analyzed by using the z statistic. Because of the confirmatory nature of this analysis, one-sided p values are reported.
Discussion
Stimulated by a series of investigations from our group (
7,
9,
11) and others (
10,
13,
15,
40) suggesting a prominent role of
FKBP5 in stress vulnerability and depression, we investigated
FKBP5 polymorphisms as promising candidates for gene-environment interactions affecting depression onset. This was done in a prospective community study with a 10-year follow-up covering the first three decades of life. In agreement with previous studies (
9–
11), we identified homozygosity for the minor allele of selected
FKBP5 polymorphisms as a risk genotype for the development of a major depressive episode in individuals with prior trauma exposure.
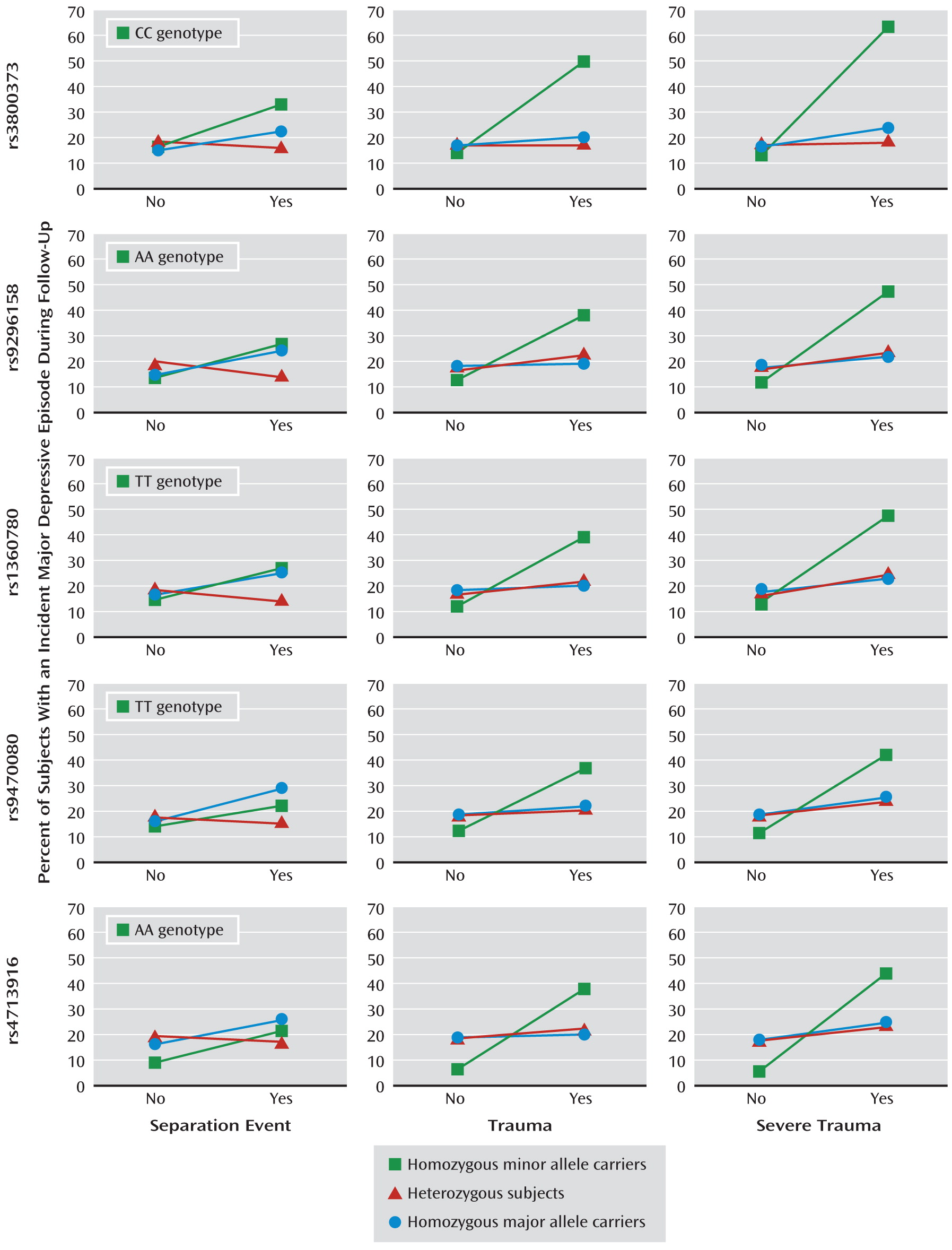
Several findings are noteworthy. First, whereas no genetic main effects for FKBP5 polymorphisms were found, severe trauma was a risk factor for depression onset. Second, consistent with a recessive model, significant interactions between the five FKBP5 SNPs and trauma indicate that subjects homozygous for the minor alleles are particularly sensitive to the pathogenic effects of trauma. The consistent effects across all five investigated polymorphisms are plausible since all SNPs are in high linkage disequilibrium. Third, while no stable interaction for separation events was revealed, there was an increasing interaction effect from trauma to severe trauma on subsequent depression onset. This dose-response relationship, together with the temporal priority, suggests a causal role of trauma exposure in this interaction process. Fourth, as variants in the five FKBP5 SNPs were not robustly associated with any of the adverse life event categories, a bias of interaction effects by gene-environment correlations is unlikely. Fifth, the interactions largely remained stable after we adjusted for controls (such as PTSD) and multiple testing. This stability indicates that the results are not confounded by these factors and further supports a causal relationship.
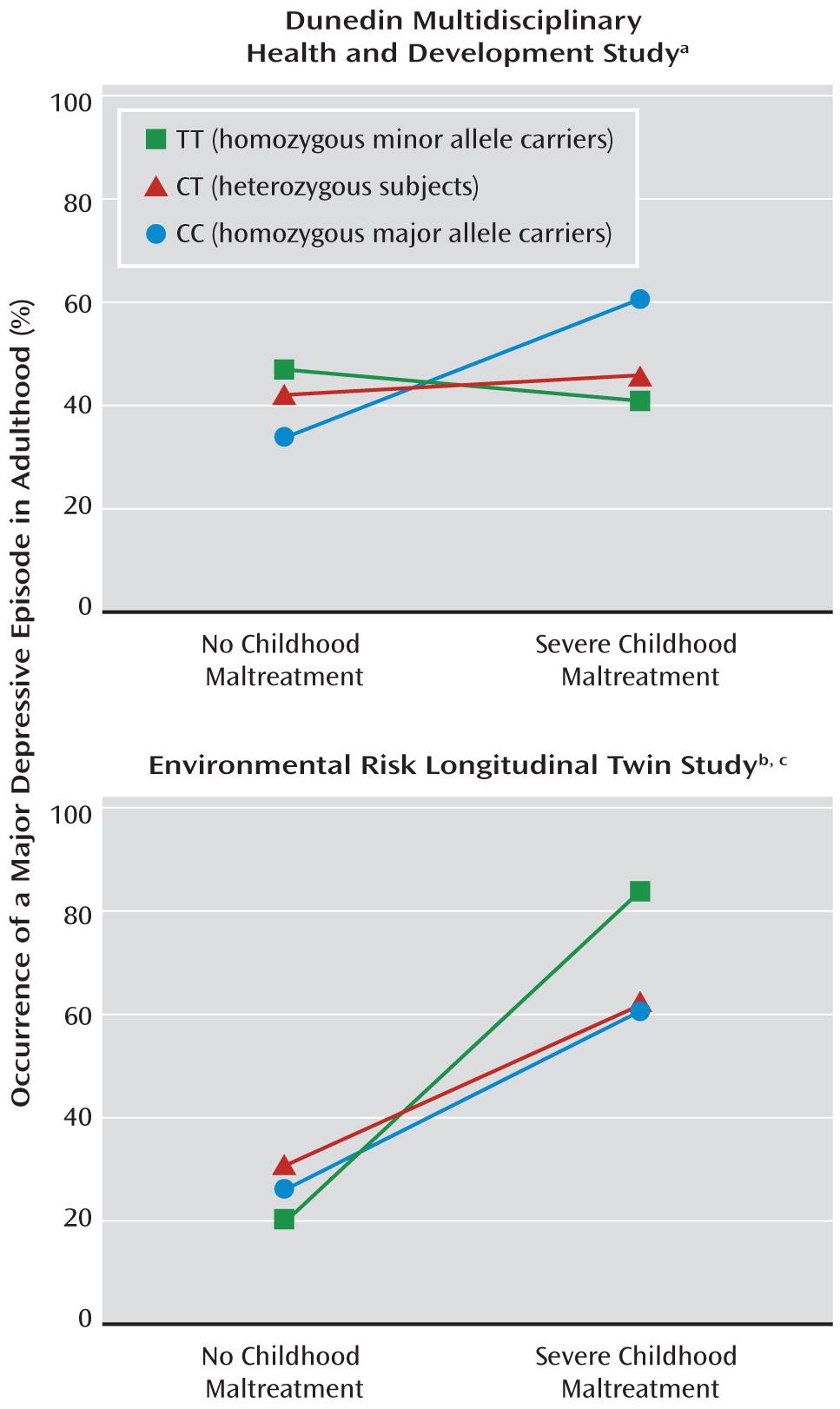
The interaction finding is supported by data from the E-Risk study: Subjects homozygous for the minor allele of the intronic FKBP5 SNP rs1360780 with exposure to severe childhood trauma showed the highest prevalence of adult major depressive episodes. No effects were found in the Dunedin study. In this study, however, exposure to adverse life events was defined in a broader fashion; in addition to self-reports, information about parent discipline style, caregiver changes, and mother-child interactions was included.
The following methodological points should be considered. First, although the analyses were based on a large community sample, the number of individuals with depressive episodes was still small for detecting interactions. However, the a priori power analysis suggested sufficient power for detecting gene-environment interactions within the limits of a small or medium effect. Second, the prospective approach required exclusion of individuals with a lifetime major depressive episode at baseline. This could have led to omission of people with depression onset shortly after trauma exposure, and thus the effect size of the association might be underestimated. Third, since adverse life events occurring after baseline were not considered in our analyses, the interpretation of our findings is restricted to traumas before age 24 and cannot be applied to traumas that occur beyond early adulthood. Fourth, because of the young age of our sample, some of the subjects without depression onset during follow-up could develop depression later in life. Such potentially false negatives may have led to more conservative results. Fifth, the SNPs of the respondents without a major depressive episode revealed a nominal deviation from the Hardy-Weinberg equilibrium. The high call rates (>99%) and further quality checks support the validity of the genotyping results (see Table ST1 in the online data supplement).
We did not find main effects of the five
FKBP5 SNPs on depression. This is in accordance with findings in some studies (
9,
17,
41) but not in others (
13–
15). The inconsistencies could be explained by the failure of these studies to consider trauma as an environmental factor. Moreover, the studies indicating a genetic main effect were based on clinical samples (
13,
14) and a sample with a high familial load for mood disorders (
15), who were probably suffering from more severe forms of depression than our community subjects. As negative life events and mood disorders are linked (
36,
42), trauma rates might tend to be higher in clinical samples, and consequently there might be a higher chance of determining genetic main effects. Furthermore, our findings are in contrast to results in a study by Lavebratt et al. (
12), who did not observe elevated depression rates among adult carriers of the TT genotype for rs1360780 who had a history of childhood problems. This discrepancy could be due to important methodological differences between our study and theirs (e.g., retrospective and different measures of childhood problems and depression, different age groups).
Regarding the underlying biological mechanisms, our results conform to those of functional studies showing a greater induction of FKBP5 expression by cortisol and, accordingly, an impaired glucocorticoid receptor sensitivity after stress in individuals who are homozygous for the minor alleles of these
FKBP5 SNPs (
9,
10). In a study with healthy subjects, we demonstrated an insufficient recovery of the cortisol secretory response to psychosocial stress in homozygous minor allele carriers of the same
FKBP5 SNPs (
11). This can be interpreted as a predisposition to chronically elevated cortisol levels in response to stress and thus may lead to long-lasting detrimental effects of chronically elevated cortisol levels. Since hypercortisolism due to an impaired regulation of the HPA axis can be regarded as a prominent feature of depression (
3,
43,
44), our result (elevated risk of major depressive episodes in trauma-exposed homozygous minor allele carriers) can be explained by direct effects of the investigated genotypes on FKBP5 expression. The increased expression might reduce glucocorticoid receptor sensitivity, subsequently impair the regulation of the HPA axis in response to stress, and therefore increase the risk for the development of a major depressive episode in trauma-exposed individuals. On the other hand, in nonexposed individuals it appears that carrying the risk genotype of the promoter SNP rs4713916 protects against the development of depression (see
Figure 2). Such opposing effects of the genotype depending on stress exposure level, which translate into a disordinal gene-environment interaction, have also been reported by Xie et al. (
20). In their participants who were homozygous for the minor allele of
FKBP5 SNP rs9470080, PTSD risk was highest for those exposed to childhood adversity and lowest for those not exposed. While functional data are still not available, it is conceivable that a hyperresponsive HPA axis might exhibit beneficial effects in the case of mild nontraumatic stress by mobilizing individual resources and enabling a sustained coping with low-level stress. In the case of traumatic stress, the negative effects of a hyperresponsive HPA axis might predominate through the induction of a chronically elevated cortisol level. It is notable, moreover, that the same risk alleles that apparently predispose a person to a major depressive episode also predict severity of PTSD symptoms when they interact with trauma (
10,
20), suggesting a shared role of
FKBP5 in the pathophysiology of these disorders. However, further prospective studies with large samples are required to provide sufficient power for a combined gene-environment analysis of both disease phenotypes.
Overall, the essential finding of our study is that trauma exposure during childhood and adolescence was a strong risk factor for developing subsequent depression in genetically vulnerable individuals, even within the extended time period of 10 years. Such medium- or long-term effects of adverse life events are explained by the model of Heim et al. (
4) proposing that a genetic predisposition together with early adverse experiences during sensitive developmental phases induce a persistent vulnerability to stress later in life. The present results suggest that this effect could be moderated by variants of the
FKBP5 gene. As it has been shown that early abuse is related to altered glucocorticoid receptor activity in adults (
45),
FKBP5 could be an important factor in such developmental processes.