Infants who later develop schizophrenia already have signs of developmental delay (
1–
9). Genes associated with schizophrenia, such as
NRG1,
ErbB4, and
DISC1, are expressed at high levels in the fetus, where they normally promote synaptogenesis (
10). Environmental risk factors associated with later expression of psychotic symptoms in offspring include maternal malnutrition (from causes ranging from famine to micronutrient deficiency), depression, anxiety, infection, and cigarette smoking (
11–
15). Abnormalities in the development of cerebral inhibitory neurons are a key pathophysiological defect in schizophrenia (
16). Cerebral inhibition develops during fetal and early postnatal life (
17,
18). Newborns whose parents are psychotic or whose mothers had depression or anxiety disorders or smoked cigarettes during pregnancy have diminished inhibition (
19). Schizophrenia and related mental illnesses are common disorders, and the genetic and perinatal environmental risk factors associated with them occur with high frequency in the general population. Thus, as for other illnesses with early developmental origins, primary prevention must take the form of a safe, population-wide intervention that promotes normal development even in healthy women and infants.
GABA, the inhibitory neurotransmitter of the mature brain, is excitatory in early fetal brain development. Expression of the membrane chloride transporter
KCC2 that switches GABA from excitatory to inhibitory is stimulated by activation of postsynaptic α7-nicotinic acetylcholine receptors (
17). α7-Nicotinic receptors are expressed at 10-fold higher levels in fetal hippocampus than in adult hippocampus, perhaps reflecting this critical developmental function, but these receptors are diminished in the hippocampal tissue of persons with schizophrenia (
20,
21). Innervation of postsynaptic α7-nicotinic receptors by cholinergic axons does not occur until the end of the third trimester (
22). Instead, they are activated by the millimolar concentrations of choline in the amniotic fluid (
23,
24).
Several maternal risk factors for schizophrenia are associated with decreased choline availability for the fetus (
24). Because of the demands for choline as a lipid component of cell membranes, the pregnant woman must increase her dietary intake of foods rich in choline, such as meats and eggs. Malnutrition may prevent that increase. Choline also may be sequestered in the mother’s liver as a result of stress from trauma, anxiety, or depression, thus depriving the fetus. Choline levels are regulated in part by phenylethanolamine methyl transferase (PEMT). Polymorphisms in the gene
PEMT associated with diminished choline levels are also associated with schizophrenia (
25).
In the inbred mouse strain DBA/2, which has genetically diminished expression of hippocampal α7-nicotinic receptors, dietary supplementation of maternal choline from conception through weaning was found to significantly increase cerebral inhibition in the adult offspring (
26). We conducted a similar randomized placebo-controlled trial of effects of perinatal choline on the development of cerebral inhibition in human infants. Perinatal choline supplementation is already advocated in the media because animal models show that it facilitates cognitive function in offspring (
27). This is the first randomized human trial conducted under a U.S. Food and Drug Administration (FDA) Investigational New Drug exemption for the purpose of ameliorating a pathophysiological deficit associated with risk for illness. Because the infants in this study were too young to show the effects of the intervention on a wider behavioral repertoire, a group of children who had been recorded electrophysiologically as infants 4 years earlier were studied to assess the behavioral sequelae of diminished infant P50 inhibition (
28).
Method
Healthy pregnant women were recruited from Denver obstetrical practices at the beginning of their second trimester. They had no self-reported illicit substance or medical marijuana use in the past 6 months, alcohol dependence, or current nicotine use (confirmed by urinary cotinine levels). Exclusion criteria included maternal history of trimethylaminuria, renal disease, liver disease, or Parkinson’s disease; maternal history of fetal death, fetal congenital malformation, fetal genetic abnormality; and current multiparous pregnancy. Women were also excluded if any fetal abnormality was revealed on initial ultrasound examination. The study was approved by the University of Colorado Institutional Review Board. Mothers were compensated for bringing their infants to the laboratory for recording.
After providing written informed consent, including the assent of the father when available, women completed a 7-day placebo lead-in. Participants whose compliance exceeded 70% by medication vial count were randomly assigned to identical-appearing placebo or drug early in the second trimester. Mothers received standard prenatal care, including standard folate and multivitamin supplements and high-resolution fetal ultrasound examinations during pregnancy. Parents’ psychiatric histories were elicited with the Structured Clinical Interview for DSM-IV Axis I Disorders. The principal diagnoses among parents were major depressive disorder, panic disorder, agoraphobia, generalized anxiety disorder, obsessive-compulsive disorder, and posttraumatic stress disorder. Presence of current psychiatric symptoms in the mother was defined as having sufficient symptoms to meet criteria for an anxiety or depression diagnosis during the pregnancy or an illness with onset prior to pregnancy with continued symptoms causing impairment during the pregnancy.
Mothers were visited at least three times by the study nurse during pregnancy to inquire about compliance with the study medication and prenatal care. Compliance was not otherwise verified. Ten of the mothers were discovered at these visits to have smoked during pregnancy, despite abstinence at enrollment. Because smoking was a prespecified exclusion criterion, their infants’ electrophysiological data were analyzed separately but were included in the safety assessments. Clinical data were acquired by the study nurse or gathered from the hospital records. The infants’ general development was checked at 6 months with the Mullen Scales of Early Learning (
29), which assess five developmental domains: 1) gross motor skills, 2) visual perception and discrimination, 3) fine motor planning and control, 4) receptive language, and 5) expressive language. An early learning composite score, derived from four of the scales (all but the gross motor subscale), estimates cognitive ability.
To assess longer-term sequelae of abnormal neonatal cerebral inhibition, we studied a cohort of 93 children who had been recruited 4 years earlier from the same population as the infants in the clinical trial (with the same inclusion and exclusion criteria) and had undergone P50 recordings as newborns (
28). We administered the Child Behavioral Checklist, a parental report of symptoms, to the mothers of 50 of these children (54%) at a mean age of 41 months.
Choline Administration as Phosphatidylcholine
Choline is catabolized by intestinal flora to trimethylurea, which has an obnoxious odor. Phosphatidylcholine cannot be catabolized, is well absorbed, and is interchangeably metabolized with choline; oral intake of phosphatidylcholine increases serum choline levels (
24). Doses of 3,600 mg each morning and 2,700 mg each evening were administered from a mean of 17.2 weeks (SD=2.1) after the mother’s last menstrual period until delivery. Phosphatidylcholine is 13%–15% choline; thus, this daily dose should provide approximately 900 mg of choline per day, about the same amount as in three large eggs. All mothers, regardless of treatment assignment, were fully advised of dietary practices that would result in similar levels of choline intake. After birth, infants received 100 mg of phosphatidylcholine in an oral suspension once daily or placebo, equivalent to the same treatment as in utero. Choline supplementation for the infant continued to 52 weeks after the mother’s last menstrual period before the pregnancy (until approximately 3 months of age) as estimated by first-trimester ultrasound. Postmortem studies have shown continually increasing
KCC2 expression over this same period (
30). The Institute of Medicine recommendation for adequate choline intake for infants is 125 mg/day (
31).
Statistical Analysis
The ratio of P50 amplitudes discriminates patients with schizophrenia from healthy subjects with larger effect size than the amplitude of either the first or second response alone or their difference (
32). However, ratios have more variance than the underlying amplitude measurements. Therefore, nonparametric analyses less affected by this variance have been used to detect genetic and treatment effects. The statistical plan established before the trial specified values ≥0.5 of the P50 ratio as indicating diminished inhibition. Values ≥0.5 for the P50 ratio have previously been associated with variants in the
CHRNA7 gene complex, including transmission of alleles that segregate with schizophrenia (
33). Analysis of the P50 ratios recorded in this study showed a significant admixture of two components, a lower one with a mean value of 0.24 (SD=0.12) and a higher one with mean value of 0.57 (SD=0.19; χ
2=8.50, df=3, p=0.037). P50 ratios ≥0.5 in this study are above the 0.98 probability distribution of the lower component, the same criterion that was used to specify an affected phenotype in previous genetic studies.
Analyses of infants’ other outcomes, including height, weight, and head circumference, were adjusted for gestation, with estimated due dates based on an early-pregnancy ultrasound.
Chi-square tests, Fisher’s exact tests, and t tests were used to compare the treatment groups. For all analyses, an alpha of 0.05 (two-tailed) was the criterion for significance. A post hoc power analysis showed that the groups included in the 1-month analysis had 85% power for determining the observed difference in primary outcome. The power to observe rare side effects was too small to be meaningful.
Results
A total of 193 women were screened, and 100 women were randomly assigned to receive choline or placebo. Seven women withdrew from the study or were lost to follow-up before the infant electrophysiological recording was made (see Figure S1 in the data supplement that accompanies the online edition of this article). Ten women were excluded from the primary outcome analysis because of smoking detected after randomization.
Safety and Tolerability of Choline Treatment
There were no significant differences in maternal outcome during pregnancy and delivery between the 46 women who received choline and the 47 who received placebo (
Table 1). The frequency of maternal side effects did not differ significantly between treatment groups. All pregnancies resulted in live births. Seven placebo-treated and four choline-treated women delivered prior to 37 weeks after their last menstrual period. There were no overall differences between groups in Apgar scores, weight, length, head circumference, and perinatal complications (
Table 2). At 1 month of age, there were still no significant differences between the two infant groups in measurements or possible side effects from the choline treatment. At 6 months, there were no differences in performance on the Mullen Scales of Early Learning.
Electrophysiological Assessment of Infants’ Inhibitory Brain Function
Electrophysiological recordings were obtained from 93 infants. Seven did not sleep or had poor quality recordings. Ten women were discovered to have smoked during the first trimester, and some were subsequently observed smoking during nursing visits during the study; their infants’ recordings were not considered in the primary analysis. Two recordings were performed, one at a mean gestation-adjusted age of 33 days, and the other at a mean adjusted age of 89 days at the termination of choline administration. Fifty-two infants received both recordings; data from all infants, whether recorded on one or both days, were analyzed. There were no differences between the choline and placebo groups (or between these two groups and participants who were excluded from the primary analysis) in maternal age, parental socioeconomic status, parental psychiatric illnesses, gestational age at birth, or infant’s sex (see Table S1 in the online data supplement). Week of treatment initiation was not significantly associated with inhibition of the auditory P50 cerebral evoked response.
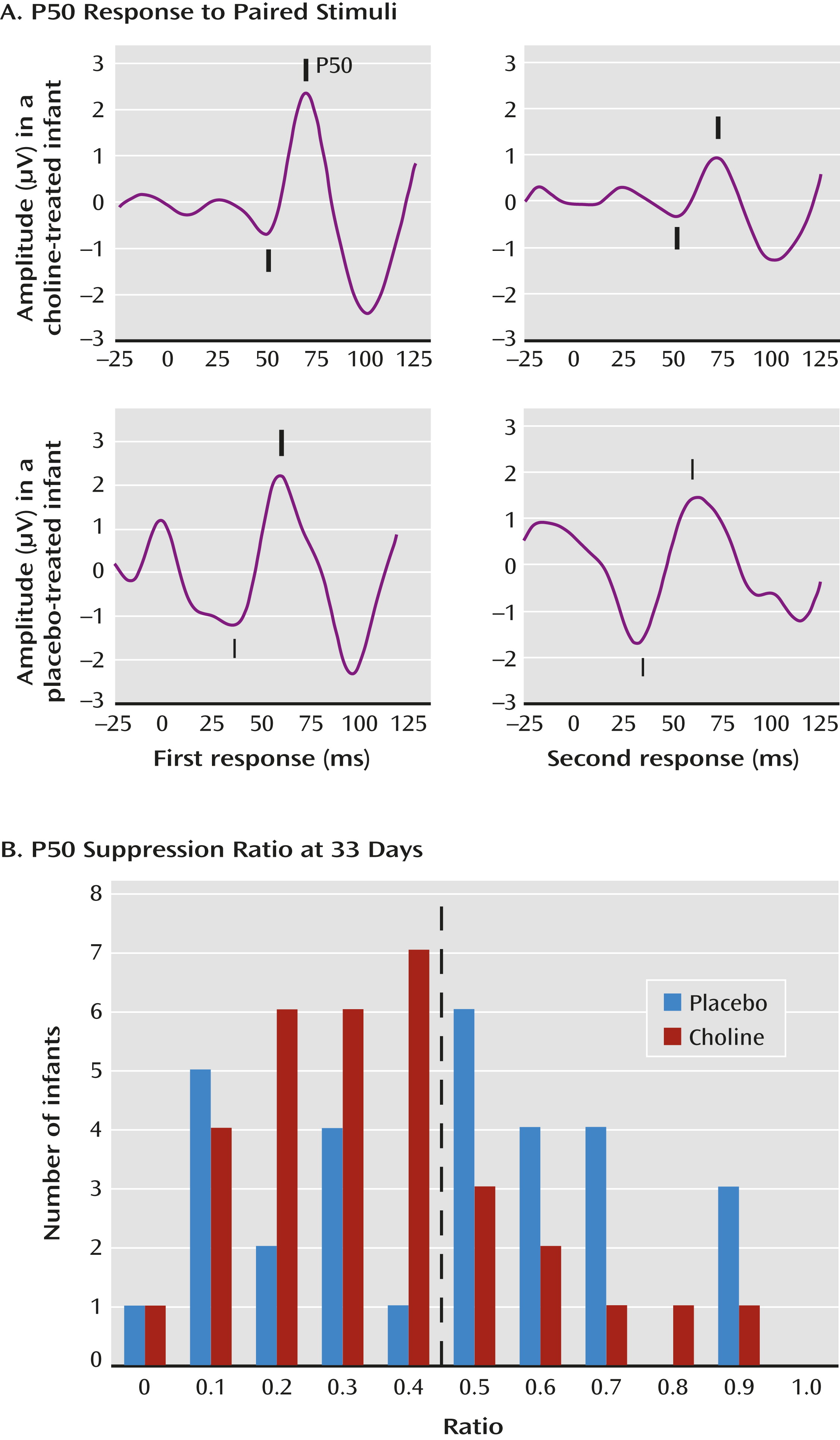
The primary outcome measure was the P50 inhibition ratio—that is, the amplitude of the P50 response to the second of paired auditory stimuli divided by the amplitude of the response to the first stimulus (
Figure 1A). A smaller ratio indicates an inhibition of the cerebral response to the repeated stimulus. The mean P50 ratio was 0.45 (SD=0.24) at age 33 days and 0.39 (SD=0.24) at age 89 days, similar to the values obtained in a previous study of 83 infants of parents with no mental illness and no tobacco exposure (
19). The a priori data analysis plan dichotomized P50 ratios into those <0.5, considered intact cerebral inhibition, and those ≥0.5, considered diminished inhibition (
Figure 1B).
At the first recording, P50 ratios <0.5 were observed in 43% of the infants treated with placebo and 76% of those treated with choline (χ2=6.90, df=1, p=0.009; d=0.7). At the second recording, 72% of the placebo-treated infants and 76% of the choline-treated infants had P50 ratios <0.5 (see Table S2 in the online data supplement).
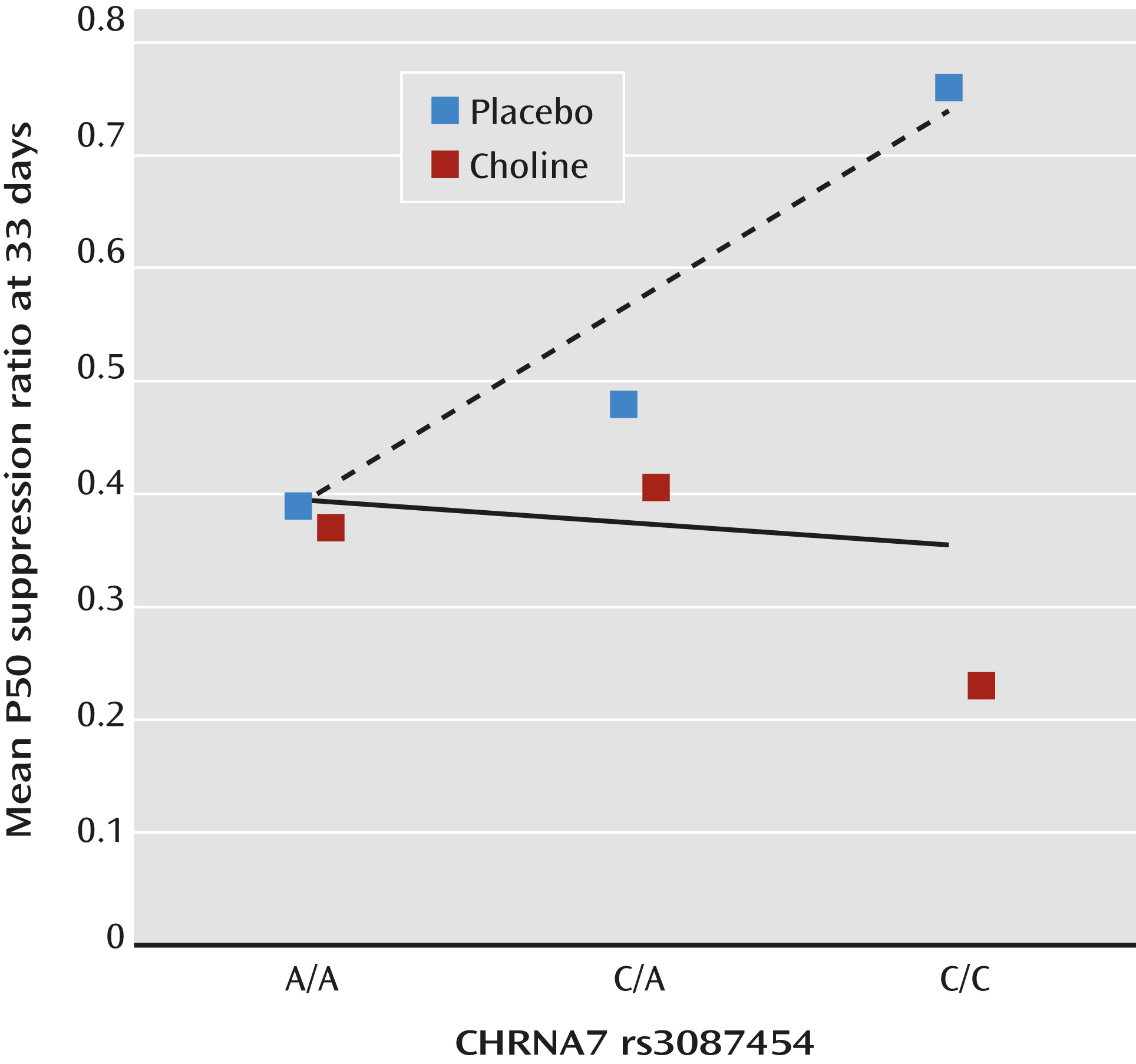
CHRNA7 rs3087454 infant genotype was correlated with P50 inhibition at 33 days in the placebo group (r
s=0.38, df=30, p=0.032), whereas the choline-treated group showed no effect (r
s=–0.05, df=28, n.s.) (
Figure 2; see also Figure S2 in the online data supplement).
Other than the ratio between the first and second P50 responses, the latencies and amplitudes of the individual P50 responses did not differ between the treatment groups. P50 ratios were correlated with the amplitudes of the P50 responses to the second stimulus (first assessment, r=0.69, p<0.001; second assessment, r=0.78, p<0.001) but not to the first stimulus (first assessment, r=0.11, p=0.375; second assessment, r=–0.09, p=0.5).
Of 10 infants whose mothers smoked during pregnancy, eight received placebo and two received choline. Postnatal smoking was not regulated. P50 recording was completed in nine of the 10 tobacco-exposed infants at the first assessment and in seven of the 10 at the second assessment. Five of the seven placebo-treated infants and both the choline-treated infants had P50 ratios <0.5 at the first recording. At the second recording, however, four of the five placebo-treated infants and one of the two choline-treated infants had P50 ratios ≥0.5. (Other subgroup analyses are summarized in Table S3 in the online data supplement).
Sequelae of Diminished Infant P50 Inhibition in Later Childhood
Among the children whose recordings from 4 years earlier indicated deficient P50 suppression in infancy, 24 had greater attentional symptoms at 3.5 years compared with 26 children with intact P50 suppression in infancy, as indicated by parent report on the Child Behavior Checklist (2.75 items [SD=1.51] compared with 1.46 items [SD=1.39]; Wilcoxon rank-sum p=0.004).
Discussion
Choline treatment of mothers during the last two trimesters of pregnancy and of their infants during the first 12 weeks of postnatal life was safely tolerated by both mother and infant. By approximately 5 postnatal weeks adjusted for duration of gestation, infants treated with choline were significantly more likely to have normal cerebral inhibition. The selection of inhibition of the auditory P50 cerebral evoked response as the primary outcome measure reflects issues in the assessment of developmental risk for schizophrenia. The primary measurement must meet four requirements: it must reflect a developmental process whose outcome is measurable in newborns, that occurs in the time window of observation and treatment, that is likely to be directly affected by the treatment, and that has a longer-term relationship to later mental illnesses. All four requirements were addressed by the inhibition of the auditory P50 cerebral evoked response. The measurement of P50 inhibition was previously shown to be feasible in human newborns (
28), the switch of GABA from an excitatory to an inhibitory neurotransmitter is a well-established fetal developmental milestone (
17,
30), the mouse model experiment showed that a similar mouse inhibition is affected by perinatal choline treatment (
26), and family and genetic studies have shown that P50 inhibition in adults and newborns is related to transmission of risk for schizophrenia (
19,
33).
Transient Developmental Delay and Risk for Later Schizophrenia
Other evidence of early developmental delay associated with later schizophrenia has been reported. A landmark behavioral observation showed dystonia and poor coordination in home films of infants who would later develop schizophrenia (
2). Recent structural MR imaging has shown differences in brain volume in male infants who have parents with schizophrenia (
34). Children who later develop schizophrenia have delayed development of attentional control as well as delayed motor, verbal, social, and general cognitive development (
1–
9). The apparent normalization of cerebral inhibition by 13 weeks in the placebo group is similar to the transient nature of other developmental delays previously observed in infants, children, and adolescents vulnerable to later psychosis (
1–
9). The apparent normalization may reflect compensatory mechanisms. Mouse
Chrna7 null mutants eventually develop GABA-mediated inhibition (
17). Maternal oxytocin released during labor, which also promotes the change in polarity of GABA receptor activation, may be one contributor to this compensatory, if delayed, developmental process (
35). Although developmental delays may be transient, the risk for later psychosis persists into adult life. In regard to cerebral inhibition, postmortem brain tissue from persons with schizophrenia continues to show only partial conversion of chloride transporters to the adult KCC2 type (
30).
Use of Surrogate Markers in the Prevention of Illness
Diminished neonatal P50 inhibition as a target for a treatment intended to prevent schizophrenia is an example of increasing efforts toward early treatment of surrogate markers for later illness. The FDA is now authorized to recognize such efforts, because early treatments directed to a surrogate marker may be the only possible strategy to prevent an illness like schizophrenia that will not appear until decades after the critical period of brain development when preventive treatment is possible (
36). The strategy has had notable successes and failures for other illnesses. Treatment of the surrogate marker high serum cholesterol prevents later heart disease. In contrast, while pioglitazone treatment of the surrogate marker glucose instability in type 2 diabetes effectively decreases the risk of later heart disease, rosiglitazone, another drug in the same class that also treats glucose instability, unexpectedly increases the risk of heart disease (
37). Both drugs bind to peroxisome proliferator-activated receptor gamma (PPARG), but their mixture of PPARG activation and inhibition was found to differ subtly. Relevant to the present study, both nicotine and choline activate α7-nicotinic receptors, but nicotine also profoundly desensitizes nicotinic receptors and choline does not (
23). Maternal smoking is associated with poorer neonatal cerebral inhibition and later childhood behavioral abnormalities, whereas choline appears to have benefits (
14,
19).
Three lessons are widely acknowledged: 1) the mechanism that links the treatment to the surrogate marker and then to the illness itself must be understood; 2) diligent efforts to understand all effects of the treatment must be made; and 3) there must be a continuing commitment to assess whether normalization of the surrogate indeed has its intended positive clinical effects on the later development of illness (36). Mechanism of the Treatment
Sensory gating deficiency assessed by P50 auditory evoked potential response to paired stimuli was initially developed as an endophenotype to assess transmission of genetic risk, which is most often through unaffected parents. P50 ratios ≥0.50 were found in 91% of unrelated patients with schizophrenia, compared with 6% of healthy comparison subjects. Generally one of two unaffected parents and about half of unaffected siblings of schizophrenia probands also had decreased P50 inhibition, and transmission of the phenotype through clinically unaffected family members to an ill proband was genetically linked with the
CHRNA7 genotype (
33). A second group of investigators replicated the genetic association between P50 gating and
CHRNA7 genotype (
38). Associations with other known schizophrenia risk genes,
COMT and
NRG1, have also been reported (
39,
40).
The mechanism of action of choline was examined in the DBA/2 mouse model by breeding a Chrna7 null mutation on the DBA/2 background. The wild-type mice, which have naturally occurring Chrna7 promoter variants responsible for partially decreased α7-nicotinic receptor expression, respond to perinatal choline treatment by developing normal hippocampal auditory evoked response inhibition. The homozygote null mutants have no α7-nicotinic receptors and fail to respond to perinatal choline treatment, demonstrating that α7-nicotinic receptor activation is the critical mechanism (41). Unlike the DBA/2 mice, the infants were not selected for CHRNA7 variants, but the genetic analysis indicates a similar genotypic effect of a CHRNA7 promoter variant on the development of cerebral inhibition in the placebo group. The placebo subjects homozygous for the minor allele associated with schizophrenia (C/C) have the highest mean ratios, those homozygous for the more common allele (A/A) have the lowest, and heterozygotes (A/C) are intermediate (Figure 2). As with the DBA/2 wild-type mice, perinatal choline treatment mitigates this genetic effect and allows infants to develop cerebral inhibition regardless of CHRNA7 genotype.
Other Actions of Choline
Choline also participates in membrane biosynthesis and one-carbon metabolic pathways, including those involved in DNA methylation (
42). However, stimulation of α7-nicotinic receptors requires higher levels of choline (K
M=300 µM) than activation by choline kinase, required to synthesize membrane sphingomyelin (K
M=17 µM), or by choline dehydrogenase, required for its role as a methyl donor (K
M=5.7 µM) (
23,
43,
44). The implication is that agonism at α7-nicotinic receptors is much more affected by supplemental dietary intake, particularly if the infant has a genetically determined receptor expression deficit.
Longer-Term Clinical Effect
A surrogate marker has never been used as a target for the prevention of a brain illness. Alzheimer’s disease, with its development over a decade, has been considered, but the development of schizophrenia over several decades, spanning development from fetal life to early adulthood, is more daunting. Therefore, the commitment to look for clinical effects is critical. P50 abnormalities appear to predict early childhood inattentiveness, as shown in the cohort initiated 4 years ago, but more such efforts, including the longer-term follow-up of children who have received supplemental choline, will be needed.
Limitations
This is the first randomized controlled trial of choline supplementation in pregnancy for any reason. There are obvious limitations. Because of the small sample in this initial trial, the frequency of most side effects cannot be determined. Nor does the trial establish whether choline supplementation will actually prevent later mental illness in the offspring. The choline dosage approximated recommended dietary levels, which would increase choline intake to twice normal levels in a mother eating an adequate diet. In the mouse experiment, we supplemented at five times dietary levels. Plasma levels were not measured, however, because they primarily reflect the most recent meal and do not reach a fasting steady state. Thus, we do not know the optimal human dosage.
Conclusions
The beneficial effects of many perinatal interventions, such as folic acid to prevent neural tube deficits, were not definitively established until they were applied population-wide (
45). The safety profile of phosphatidylcholine would support such administration. Schizophrenia, with its low rate (10%) of parent-to-child transmission and the high population allele frequencies of most risk genes, including
CHRNA7, merits population-wide prevention, but determining the ultimate outcome of the choline intervention represents a formidable problem because the illness itself does not appear until decades later. Changes in biomarkers such as P50 inhibition in the neonatal period in studies such as this one are thus the only available indicator of the putative success of intervention in the most vulnerable developmental period.
Acknowledgments
The authors thank Drs. Steve Zeisel and Gary O. Zerbe for their help in study design and Drs. William Hay, Henry Galan, Brian Stafford, and Kim Kelsay for their involvement in the data safety monitoring board.