An 8-Week Randomized, Double-Blind, Placebo-Controlled Evaluation of the Safety and Efficacy of Cariprazine in Patients With Bipolar I Depression
Abstract
Objective:
Method:
Results:
Conclusions:
Method
Study Design
Patients
Efficacy Evaluations
Safety Evaluations
Statistical Analysis
Results
Patient Disposition and Demographic Characteristics
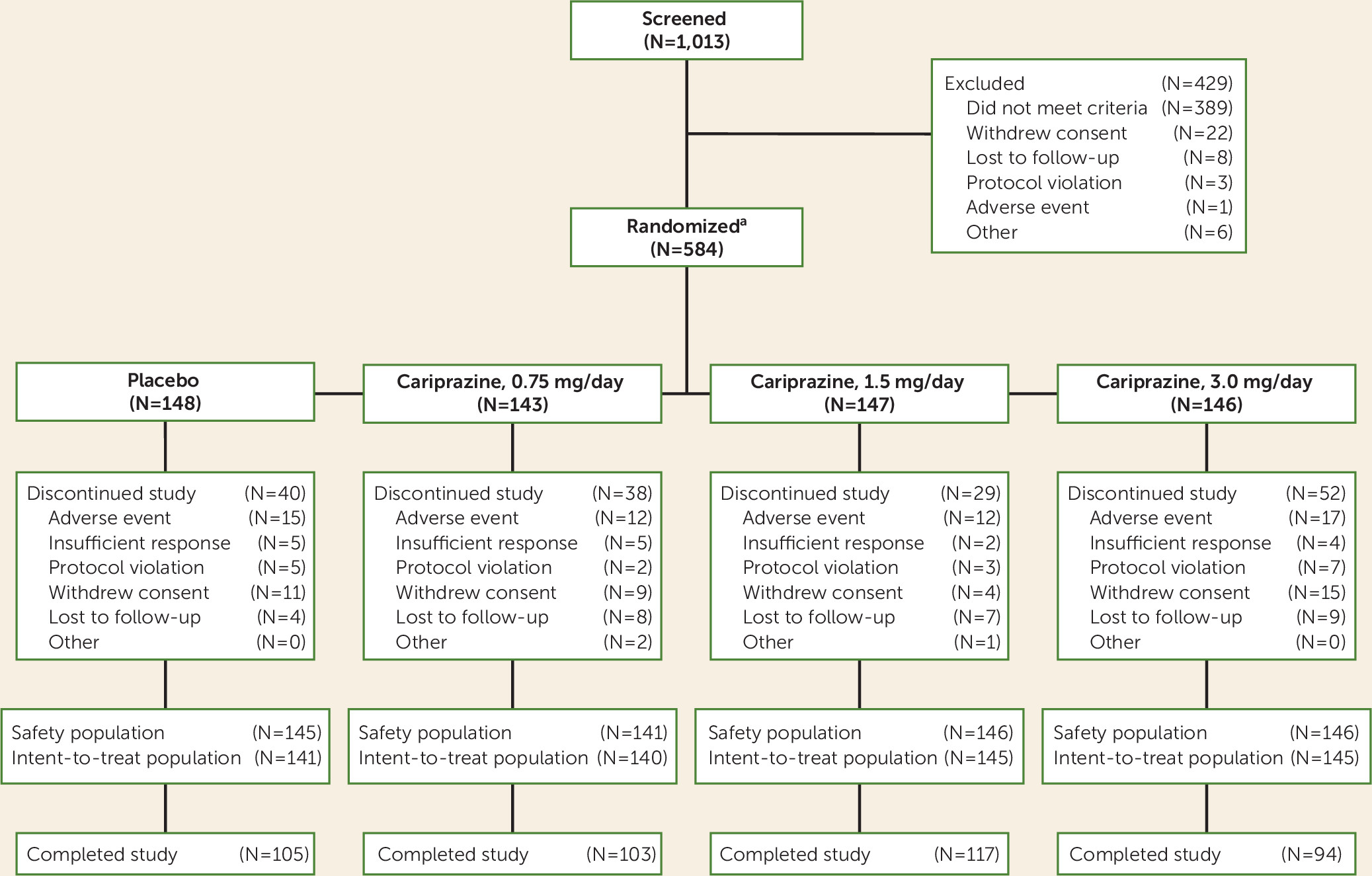
| Cariprazine | ||||||||
|---|---|---|---|---|---|---|---|---|
| Characteristic | Placebo | 0.75 mg/day | 1.5 mg/day | 3.0 mg/day | ||||
| N | % | N | % | N | % | N | % | |
| Female | 89 | 61.4 | 91 | 64.5 | 92 | 63.0 | 88 | 60.3 |
| Race | ||||||||
| White | 110 | 75.9 | 111 | 78.7 | 109 | 74.7 | 113 | 77.4 |
| Black or African American | 30 | 20.7 | 26 | 18.4 | 30 | 20.5 | 26 | 17.8 |
| Other | 5 | 3.5 | 4 | 2.8 | 7 | 4.8 | 7 | 4.8 |
| Mean | SD | Mean | SD | Mean | SD | Mean | SD | |
| Age (years)a | 43.6 | 12.0 | 40.1 | 11.2 | 40.9 | 11.4 | 42.8 | 10.8 |
| Weight (kg) | 80.0 | 17.1 | 80.8 | 18.4 | 81.4 | 16.8 | 81.5 | 17.9 |
| BMI | 27.8 | 5.3 | 28.4 | 5.7 | 28.4 | 5.4 | 28.3 | 5.6 |
| Age at onset of original episode (years) | 28.4 | 11.4 | 26.0 | 10.2 | 25.4 | 10.2 | 28.1 | 11.0 |
| Duration of bipolar disorder (years) | 15.3 | 10.2 | 14.1 | 9.7 | 15.5 | 10.3 | 14.6 | 9.5 |
| Duration of current depressive episode (months) | 3.3 | 2.3 | 3.8 | 2.6 | 3.7 | 2.7 | 3.5 | 2.4 |
| Number of lifetime depressive episodes | 6.2 | 5.8 | 5.7 | 5.2 | 7.2 | 8.0 | 6.8 | 7.0 |
| Measure, Model, and Group | Analyses | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Baseline score | Change | Difference versus placebo | |||||||
| N | Mean | SD | LS mean | SE | LSMD | 95% CI | p | Adjusted p | |
| Primary Efficacy Parameter: MADRS | |||||||||
| Mixed-effects model for repeated measures | |||||||||
| Placebo | 141 | 30.4 | 4.6 | –11.1 | 0.9 | ||||
| Cariprazine 0.75 mg/day | 140 | 31.1 | 4.7 | –13.0 | 0.9 | –1.9 | –4.3, 0.5 | 0.129 | 0.129 |
| Cariprazine 1.5 mg/day | 145 | 30.3 | 4.4 | –15.1 | 0.8 | –4.0 | –6.3, –1.6 | 0.001 | 0.003 |
| Cariprazine 3.0 mg/day | 145 | 30.6 | 4.7 | –13.7 | 0.9 | –2.5 | –4.9, –0.1 | 0.037 | 0.112 |
| ANCOVA with last observation carried forward | |||||||||
| Placebo | 141 | 30.4 | 4.6 | –10.1 | 0.8 | ||||
| Cariprazine 0.75 mg/day | 140 | 31.1 | 4.7 | –12.4 | 0.8 | –2.3 | –4.6, –0.1 | 0.041 | |
| Cariprazine 1.5 mg/day | 145 | 30.3 | 4.4 | –14.2 | 0.8 | –4.1 | –6.3, –1.9 | <0.001 | |
| Cariprazine 3.0 mg/day | 145 | 30.6 | 4.7 | –12.8 | 0.8 | –2.7 | –4.9, –0.5 | 0.017 | |
| Secondary Efficacy Parameter: CGI-S | |||||||||
| Mixed-effects model for repeated measures | |||||||||
| Placebo | 141 | 4.4 | 0.5 | –1.0 | 0.1 | ||||
| Cariprazine 0.75 mg/day | 140 | 4.4 | 0.5 | –1.1 | 0.1 | –0.1 | –0.4, 0.1 | 0.303 | 0.303 |
| Cariprazine 1.5 mg/day | 145 | 4.4 | 0.5 | –1.4 | 0.1 | –0.4 | –0.6, –0.1 | 0.004 | 0.013 |
| Cariprazine 3.0 mg/day | 145 | 4.4 | 0.5 | –1.3 | 0.1 | –0.3 | –0.5, –0.0 | 0.049 | 0.112 |
| ANCOVA with last observation carried forward | |||||||||
| Placebo | 141 | 4.4 | 0.5 | –0.9 | 0.1 | ||||
| Cariprazine 0.75 mg/day | 140 | 4.4 | 0.5 | –1.0 | 0.1 | –0.2 | –0.4, 0.1 | 0.198 | |
| Cariprazine 1.5 mg/day | 145 | 4.4 | 0.5 | –1.3 | 0.1 | –0.4 | –0.6, –0.2 | 0.001 | |
| Cariprazine 3.0 mg/day | 145 | 4.4 | 0.5 | –1.2 | 0.1 | –0.3 | –0.5, –0.0 | 0.024 | |
| Additional Efficacy Parameters | |||||||||
| HAM-D | |||||||||
| Mixed-effects model for repeated measures | |||||||||
| Placebo | 141 | 24.1 | 2.8 | –9.1 | 0.6 | ||||
| Cariprazine 0.75 mg/day | 140 | 24.6 | 3.4 | –10.3 | 0.6 | –1.1 | –2.9, 0.6 | 0.199 | |
| Cariprazine 1.5 mg/day | 145 | 23.9 | 3.2 | –11.8 | 0.6 | –2.7 | –4.4, –1.0 | 0.002 | |
| Cariprazine 3.0 mg/day | 145 | 24.0 | 3.1 | –11.3 | 0.6 | –2.2 | –3.9, –0.5 | 0.013 | |
| ANCOVA with last observation carried forward | |||||||||
| Placebo | 141 | 24.1 | 2.8 | –8.4 | 0.6 | ||||
| Cariprazine 0.75 mg/day | 140 | 24.6 | 3.4 | –9.7 | 0.6 | –1.4 | –3.1, 0.3 | 0.098 | |
| Cariprazine 1.5 mg/day | 145 | 23.9 | 3.2 | –11.2 | 0.6 | –2.9 | –4.5, –1.2 | 0.001 | |
| Cariprazine 3.0 mg/day | 145 | 24.0 | 3.1 | –10.6 | 0.6 | –2.2 | –3.9, –0.6 | 0.007 | |
| Odds Ratio Versus Placebo | |||||||||
| n | % | Odds Ratio | 95% CI | p | |||||
| Response at week 6 (≥50% score reduction on MADRS)b | |||||||||
| Placebo | 45 | 31.9 | |||||||
| Cariprazine 0.75 mg/day | 54 | 38.6 | 1.35 | 0.83, 2.22 | 0.227 | ||||
| Cariprazine 1.5 mg/day | 72 | 49.7 | 2.10 | 1.30, 3.41 | 0.002 | ||||
| Cariprazine 3.0 mg/day | 65 | 44.8 | 1.74 | 1.07, 2. 82 | 0.024 | ||||
| Remission at week 6 (score ≤10 on MADRS)b | |||||||||
| Placebo | 28 | 19.9 | |||||||
| Cariprazine 0.75 mg/day | 33 | 23.6 | 1.32 | 0.74, 2.36 | 0.340 | ||||
| Cariprazine 1.5 mg/day | 53 | 36.6 | 2.38 | 1.38, 4.09 | 0.002 | ||||
| Cariprazine 3.0 mg/day | 40 | 27.6 | 1.59 | 0.91, 2.78 | 0.105 | ||||
| HAM-D remitters (score ≤7)b | |||||||||
| Placebo | 22 | 15.6 | |||||||
| Cariprazine 0.75 mg/day | 28 | 20.0 | 1.44 | 0.77, 2.67 | 0.254 | ||||
| Cariprazine 1.5 mg/day | 44 | 30.3 | 2.34 | 1.31, 4.18 | 0.004 | ||||
| Cariprazine 3.0 mg/day | 31 | 21.4 | 1.46 | 0.80, 2.69 | 0.219 | ||||
Efficacy Outcomes
Primary, secondary, and additional efficacy outcomes.
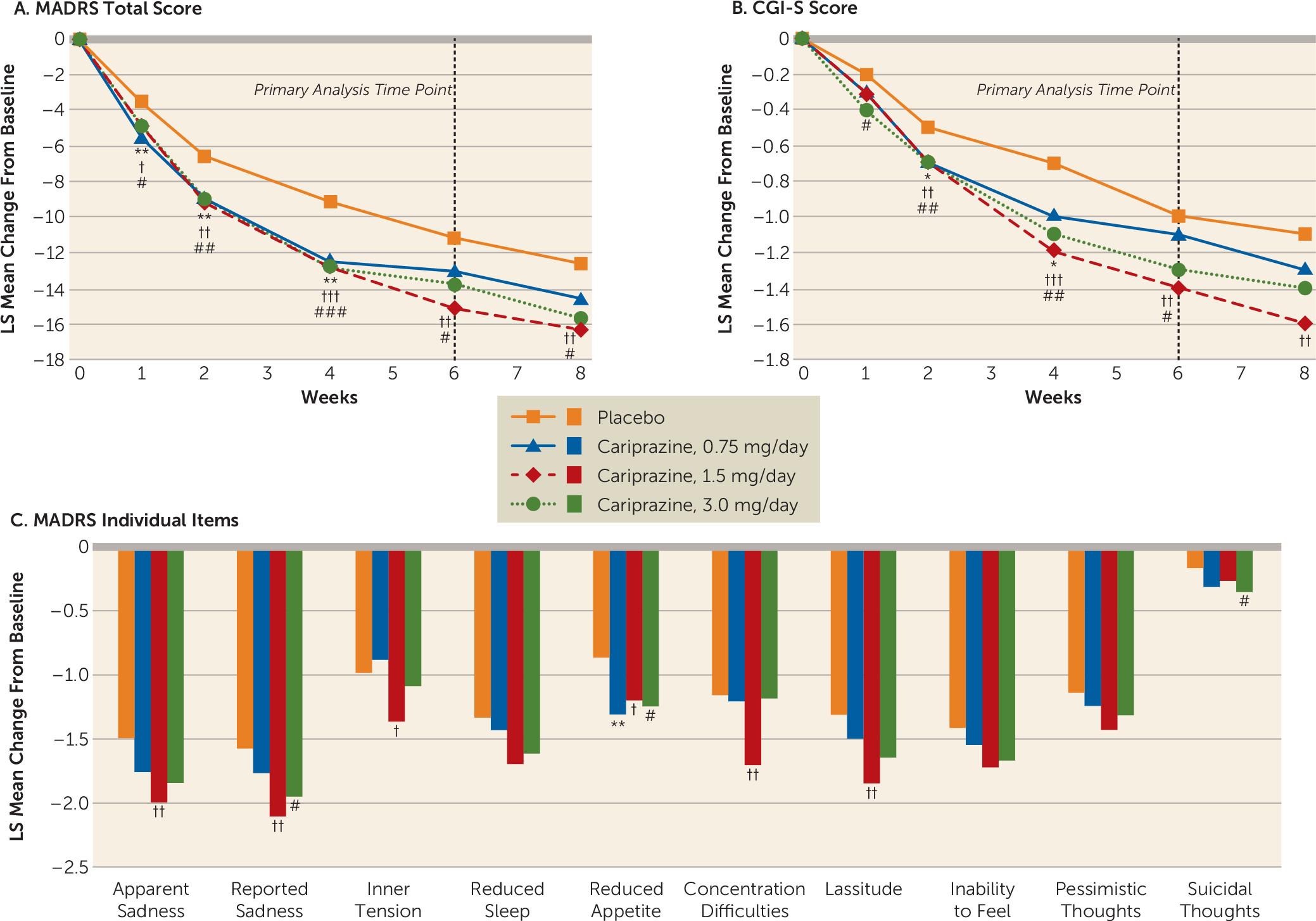
Post hoc efficacy analyses.
Safety Outcomes
Extent of exposure.
Adverse events.
| Cariprazine | ||||||||
|---|---|---|---|---|---|---|---|---|
| Placebo (N=145) | 0.75 mg/day (N=141) | 1.5 mg/day (N=146) | 3.0 mg/day (N=146) | |||||
| Measure | N | % | N | % | N | % | N | % |
| Overall adverse event summary | ||||||||
| Patients with any treatment-emergent adverse event | 79 | 54.5 | 80 | 56.7 | 91 | 62.3 | 91 | 62.3 |
| Patients with serious adverse event | 5 | 3.4 | 1 | 0.7 | 2 | 1.4 | 2 | 1.4 |
| Deaths | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 |
| Patients who discontinued due to adverse events | 15 | 10.3 | 12 | 8.5 | 12 | 8.2 | 17 | 11.6 |
| Patients with newly emergent adverse eventsa | 6 | 4.1 | 2 | 1.4 | 5 | 3.4 | 3 | 2.1 |
| Adverse events during double-blind treatment period (≥5% in any treatment group) | ||||||||
| Akathisia | 2 | 1.4 | 4 | 2.8 | 7 | 4.8 | 21 | 14.4 |
| Insomnia | 12 | 8.3 | 16 | 11.3 | 10 | 6.8 | 17 | 11.6 |
| Nausea | 7 | 4.8 | 12 | 8.5 | 12 | 8.2 | 12 | 8.2 |
| Headache | 16 | 11.0 | 11 | 7.8 | 10 | 6.8 | 10 | 6.8 |
| Somnolence | 6 | 4.1 | 6 | 4.3 | 9 | 6.2 | 10 | 6.8 |
| Restlessness | 5 | 3.4 | 4 | 2.8 | 4 | 2.7 | 9 | 6.2 |
| Diarrhea | 8 | 5.5 | 2 | 1.4 | 9 | 6.2 | 3 | 2.1 |
| Irritability | 1 | 0.7 | 7 | 5.0 | 3 | 2.1 | 2 | 1.4 |
Laboratory values, vital signs, and other safety parameters.
| Cariprazine | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Parameter | Placebo | 0.75 mg/day | 1.5 mg/day | 3.0 mg/day | ||||||||
| N | Mean | SD | N | Mean | SD | N | Mean | SD | N | Mean | SD | |
| Liver function | ||||||||||||
| ALT (U/L) | 131 | 0.4 | 11.1 | 120 | 0.1 | 16.8 | 132 | 1.4 | 9.4 | 125 | 1.7 | 21.6 |
| AST (U/L) | 131 | –0.8 | 7.1 | 120 | –0.1 | 11.1 | 132 | 0.6 | 7.1 | 124 | 0.2 | 6.9 |
| Total bilirubin (mg/dL) | 131 | –0.03 | 0.24 | 119 | –0.01 | 0.20 | 132 | 0.00 | 0.24 | 125 | –0.02 | 0.25 |
| Metabolic parameters | ||||||||||||
| Cholesterol | ||||||||||||
| HDL (mg/dL) | 131 | 0.73 | 13.69 | 120 | 0.88 | 12.10 | 132 | 0.46 | 10.33 | 124 | –1.64 | 9.49 |
| LDL (mg/dL)a | 131 | –3.41 | 23.48 | 120 | –2.78 | 21.96 | 132 | –3.18 | 23.40 | 124 | –6.40 | 23.86 |
| Total (mg/dL) | 131 | –2.50 | 30.12 | 120 | –3.93 | 29.13 | 132 | –2.56 | 28.83 | 125 | –6.76 | 27.00 |
| Triglycerides (mg/dL) | 115 | 1.97 | 64.38 | 105 | –5.27 | 90.14 | 119 | –2.36 | 61.26 | 106 | 7.51 | 74.44 |
| Fasting glucose (mg/dL) | 115 | 3.63 | 13.91 | 105 | 1.97 | 18.90 | 119 | 1.45 | 13.17 | 106 | 8.08 | 20.31 |
| Chemistry parameters | ||||||||||||
| Prolactin (ng/mL) | 131 | –1.00 | 15.50 | 119 | 0.59 | 8.35 | 131 | 0.25 | 14.29 | 123 | 1.54 | 15.22 |
| Creatine kinase (U/L) | 131 | –2.3 | 94.9 | 120 | 13.0 | 66.2 | 132 | 27.3 | 191.6 | 124 | 9.4 | 70.8 |
| Vital signs | ||||||||||||
| Blood pressureb | ||||||||||||
| Systolic (mmHg) | 142 | –0.7 | 10.4 | 140 | –1.4 | 8.3 | 145 | 1.3 | 8.7 | 145 | –0.6 | 12.0 |
| Diastolic (mmHg) | 142 | –0.1 | 8.0 | 140 | –0.7 | 7.8 | 145 | 0.8 | 6.5 | 145 | –0.4 | 7.5 |
| Pulseb | 142 | 0.2 | 10.4 | 140 | –0.6 | 10.0 | 145 | 1.4 | 10.8 | 145 | 2.2 | 10.2 |
| Body weight (kg) | 142 | 0.10 | 2.28 | 140 | 0.84 | 2.50 | 145 | 1.10 | 2.64 | 145 | 0.63 | 2.26 |
| Waist circumference (cm) | 142 | 0.08 | 4.07 | 140 | 0.77 | 3.48 | 145 | 0.23 | 3.33 | 145 | 0.53 | 3.54 |
| N | n | % | N | n | % | N | n | % | N | n | % | |
| Other safety outcomes | ||||||||||||
| Orthostatic hypotensionc | 142 | 17 | 12.0 | 139 | 19 | 13.7 | 145 | 12 | 8.3 | 143 | 13 | 9.1 |
| Treatment-emergent parkinsonismd | 142 | 0 | 0 | 140 | 2 | 1.4 | 145 | 2 | 1.4 | 145 | 3 | 2.1 |
| Treatment-emergent akathisiae | 142 | 6 | 4.2 | 140 | 8 | 5.7 | 145 | 12 | 8.3 | 145 | 25 | 17.2 |
| Treatment-emergent maniaf | 142 | 5 | 3.5 | 140 | 6 | 4.3 | 145 | 4 | 2.8 | 145 | 4 | 2.8 |
Suicidality.
Discussion
Acknowledgments
Footnotes
Supplementary Material
- View/Download
- 189.27 KB
References
Information & Authors
Information
Published In


History
Authors
Funding Information
Metrics & Citations
Metrics
Citations
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
For more information or tips please see 'Downloading to a citation manager' in the Help menu.
View Options
View options
PDF/EPUB
View PDF/EPUBLogin options
Already a subscriber? Access your subscription through your login credentials or your institution for full access to this article.
Personal login Institutional Login Open Athens loginNot a subscriber?
PsychiatryOnline subscription options offer access to the DSM-5-TR® library, books, journals, CME, and patient resources. This all-in-one virtual library provides psychiatrists and mental health professionals with key resources for diagnosis, treatment, research, and professional development.
Need more help? PsychiatryOnline Customer Service may be reached by emailing [email protected] or by calling 800-368-5777 (in the U.S.) or 703-907-7322 (outside the U.S.).