Major depressive disorder is the second leading cause of disability worldwide and affects an estimated 350 million people (
1). Although strides have been made in the treatment of depression, an estimated 15% of depressed individuals do not respond despite adequate pharmacotherapy, psychotherapy, and even ECT (
2). Current predictors of treatment response for depression, such as severity, chronicity, and exposure to adverse life events, are nonspecific and without clear therapeutic implications in treatment-refractory depression. The clinical management of treatment-refractory depression has not advanced significantly in the past two decades, and the suicide rate in the United States has been increasing (
3). Recent emphasis on treatment-refractory depression has focused on neuromodulatory treatments, such as deep brain stimulation, which, while promising, requires neurosurgical intervention. We propose a novel diagnostic and therapeutic approach to treatment-refractory depression based on a targeted analysis of metabolites in blood, urine, and CSF.
Metabolomics is the study of patterns of metabolic intermediates to characterize dysfunctional metabolic pathways potentially related to symptomatic presentation. This approach has previously contributed to our understanding of the pathophysiology of depression. For example, over three decades ago, decreased CSF levels of 5-hydroxyindoleacetic acid (5-HIAA) in depressed patients were shown to be associated with a markedly elevated risk of suicide (
4). This work helped spur the development of selective serotonin reuptake inhibitors, which are now widely used for the treatment of depression (
5,
6). In addition, neuropsychiatric symptoms are a common manifestation of many inborn errors of metabolism. Adolescent and adult neuropsychiatric symptoms, possibly caused by metabolic abnormalities, are also a common comorbidity in both primary (
7) and secondary forms of mitochondrial dysfunction (
8).
We previously reported on the case of a 19-year-old male patient with unremitting treatment-refractory depression and repeated suicide attempts who had deficient CSF levels of tetrahydrobiopterin intermediates and responded to replacement with sapropterin, a tetrahydrobiopterin analogue (
9). CSF testing of five additional adolescent patients with treatment-refractory depression revealed that three had low CSF levels of 5-methyltetrahydrofolate (5-MTHF) and normal blood levels of folate, indicating cerebral folate deficiency (
10). None of the conventional clinical or research diagnostic or therapeutic approaches would have identified the source of these patients’ difficulties, nor would they have suggested the subsequent successful treatment strategies.
We hypothesized that the incidence of metabolic abnormalities contributing to treatment-refractory depression would be significantly greater than in healthy comparison subjects. Broader identification of such metabolic disorders could transform psychiatric practice, particularly in severe depression, when standard treatments are ineffective. We describe the systematic evaluation of 33 patients with treatment-refractory depression for primary and secondary disorders of CNS metabolism. Targeted studies of CSF and peripheral samples (blood and urine) were utilized to identify patients with abnormalities in the monoamine neurotransmitter synthesis pathway, as well other metabolomic abnormalities leading to apparent isolated CNS dysfunction with depression.
Method
Sample Characteristics
Participants 14–40 years of age with depression that has not responded to at least three maximum-dose medication trials of at least 6 weeks each were recruited by advertisement through the Clinical and Translational Science Institute’s Research Participant Registry at the University of Pittsburgh or by clinical referral. All adult participants provided informed consent; adolescent participants provided informed assent for involvement in the study and parents provided informed consent. We compared this group with young adult healthy comparison subjects who had no personal or first-degree-relative history of psychiatric disorder or suicidal behavior. Adolescent comparison subjects were not included because lumbar puncture confers greater than minimal risk. The study was approved by the Institutional Review Board of the University of Pittsburgh.
A total of 38 patients with treatment-refractory depression and 22 healthy comparison subjects were enrolled. One comparison subject was subsequently excluded after discovery of substance use disorder and suicidal behavior in one of the participant’s parents and another after discovery of self-administration of large quantities of B vitamins. One participant in the depression group was excluded because of a diagnosis of Asperger’s syndrome. Four participants in the depression group and four in the comparison group were excluded because they declined lumbar puncture.
Participants were assessed with a structured psychiatric interview, including the Family Interview for Genetics Studies (
11), at the time of referral to characterize depression course, comorbidity, family history, and history of trauma, psychosis, and anxiety. Treatment-refractory depression was confirmed with the Antidepressant Treatment History Questionnaire (
12). All participants completed the Beck Depression Inventory (BDI) (
13) and the Suicidal Ideation Questionnaire (
14), and for adolescents, the Child Depression Rating Scale–Revised (
15) was completed on the basis of clinical assessment. Patients remained on their current medications and other treatments during the course of the study.
Study Procedures
The research staff and principal investigator (L.P.) completed structured psychiatric interviews, administered self-report symptom questionnaires, and arranged for study laboratory testing. Assessment consisted of a psychiatric interview, review of records, and self-report instruments at intake to characterize depression course (BDI), suicidal ideation and behavior (Suicidal Ideation Questionnaire, Columbia Suicide History Form [
16], and Beck Suicide Ideation Scale [
17]), comorbidity (anxiety, psychosis, substance use, attention disorders, assessed with the DSM-5 Level 1 Cross-Cutting Symptom Measure), and family history (Family Interview for Genetic Studies and three-generation pedigree) (
11). A neurologic examination was conducted by the principal investigator.
Testing for CNS-specific metabolic abnormalities included 5-methyltetrahydrofolate, tetrahydrobiopterin, neopterin, pyridoxal-5-phosphate, 5-hydroxyindoleacetic acid, homovanillic acid, and amino-adipic semialdehyde. Analysis of blood and CSF began immediately after collection. Plasma and urine testing were performed by the clinical biochemical genetics and clinical chemistry laboratories of University of Pittsburgh Medical Center (UPMC), per their standard protocols, including gas chromatography-mass spectrometry of urine and liquid chromatography tandem mass spectrometry and high-performance liquid chromatography tandem mass spectrometry profiling of blood to examine groups of metabolites known to contribute to depression. Lumbar puncture for CSF collection was performed under fluoroscopic guidance by the Neuroradiology Department at UPMC, and samples were analyzed as for blood and urine by the clinical biochemical genetics and clinical chemistry laboratories of UPMC and Medical Neurogenetics, Inc. (Atlanta). If a specific abnormality was suspected on the basis of the initial testing, the patient was referred to a biochemical geneticist for additional confirmatory testing. Once results of testing became available, a follow-up appointment was scheduled for all participants in the depression group to review results and provide additional referrals if needed. At this appointment, the BDI, the Suicidal Ideation Questionnaire, and the DSM-5 Level 1 Cross-Cutting Symptom Measure were repeated. Participants for whom a novel treatment was available were recontacted at least 6 weeks after start of treatment to determine outcome with repeated administration of the BDI, the Suicidal Ideation Questionnaire, and the DSM-5 Level 1 Cross-Cutting Symptom Measure.
Analyses were conducted in IBM SPSS Statistics, version 21 (IBM, Armonk, N.Y.). We employed independent t tests to examine the differences between the depression and healthy comparison groups in depression severity (BDI), suicidal ideation (Suicidal Ideation Questionnaire), and age. A chi-square goodness-of-fit test was used to compare the sex distribution of each group. Because of the small number of participants who were assessed before and after intervention (N=10), Wilcoxon signed-ranks tests were performed between pre- and posttreatment BDI and Suicidal Ideation Questionnaire scores among the participants who were deficient in cerebral folate.
Discussion
To our knowledge, this CNS-specific metabolomic survey is the first to systematically evaluate abnormalities of neurotransmitter, vitamin, pterin, and energy metabolism in peripheral and CSF samples from patients with isolated psychiatric symptoms in the absence of primary neurologic symptoms. It was spurred by our initial identification of a young man with a severe deficiency of CSF tetrahydrobiopterin who responded to treatment with sapropterin, followed by our discovery of CFD in three of five additional patients (
9). In this case-control study of 33 individuals with treatment-refractory depression, 21 had metabolite abnormalities. Results were determined based on established clinical norms for all tests reported.
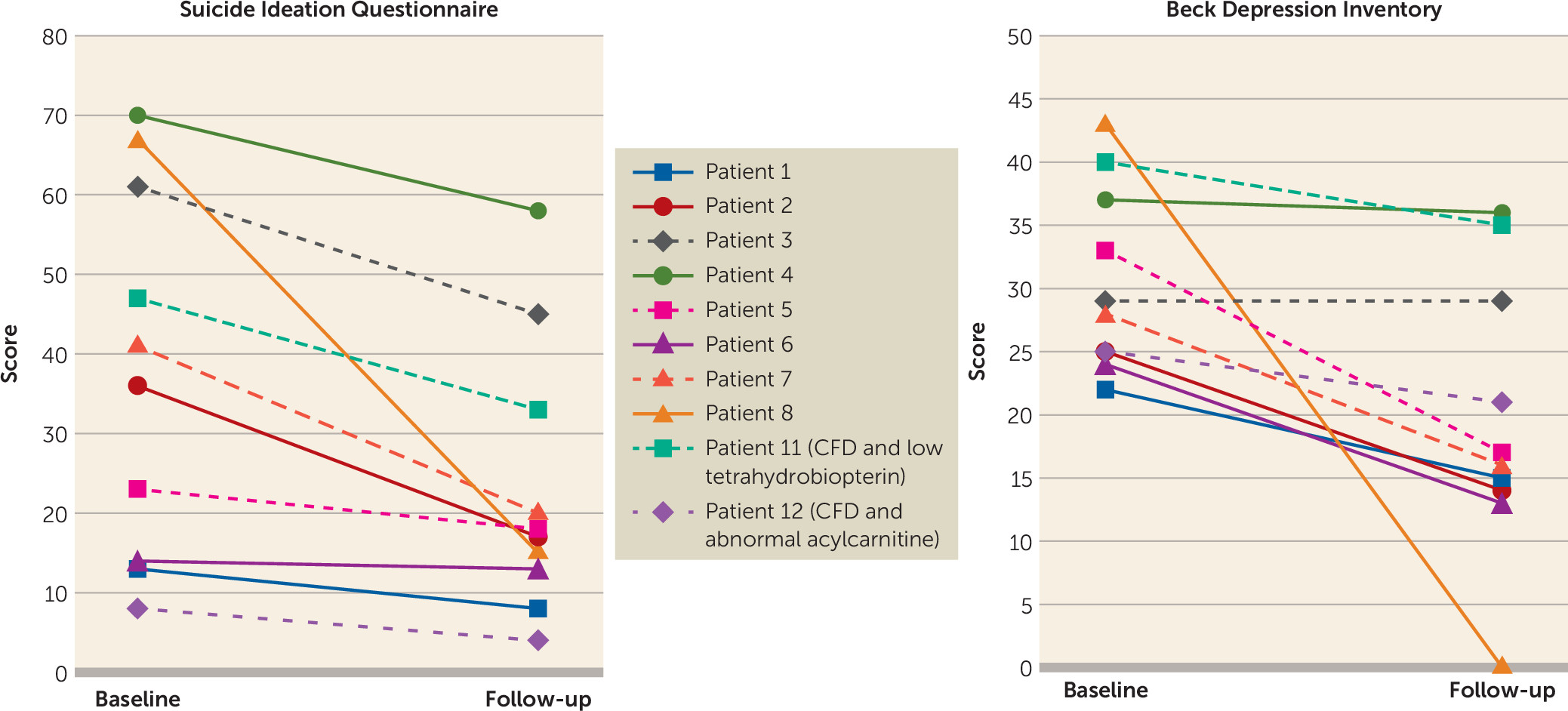
Twelve patients had CFD, and in 10 of these patients BDI and Suicidal Ideation Questionnaire scores were reduced after treatment with folinic acid; of the remaining two patients, one was nonadherent with folinic acid treatment and the other was lost to follow-up. Half of the eight patients who completed follow-up and did not have a comorbid identified metabolic abnormality had reductions in BDI score to below the threshold for depression. In all cases, patients had normal serum metabolites associated with folate pathways and therefore would have eluded conventional diagnostic approaches without a more comprehensive evaluation.
It should be noted that the abnormalities identified may be due either to a primary genetic disorder in a metabolic pathway or a secondary abnormality of indirect etiology (e.g., injury to the CNS from infection, autoimmune disease, asphyxia, or epilepsy), typically with other obvious systemic manifestations (
18). The abnormalities identified could also be sequelae of unremitting depression. CFD is a CNS-specific syndrome characterized by low CSF 5-MTHF levels and normal plasma folate levels (
19,
20). It can be caused primarily by mutations in the cerebral folate receptor gene (
FOLR1) but can be secondarily reduced in any metabolic defect that increases use of methyl groups, or by changes in transport across the blood-brain barrier.
FOLR1 gene sequencing was normal in the first eight patients with identified CFD. Secondary CFD is also seen in disorders of the mitochondrial respiratory chain, such as Alpers’ syndrome (
21), Kearns-Sayre syndrome (
22), and others (
23). Folate is involved in nearly 100 metabolic reactions (
24), including the purine synthetic pathway, contributing to impaired tetrahydrobiopterin, serotonin, norepinephrine, and dopamine synthesis, and thus polygenic effects due to the accumulation of mutations in multiple genes may play a role in this population (
25,
26).
The findings in this patient population are different from those in reports of empirical treatment with
l-methylfolate (
27–
30). Adjunctive treatment with
l-methylfolate may improve depressive symptoms because of its role as a cofactor in monoamine metabolism (
31). This is different from our findings in CFD (
18). Also,
l-methylfolate is most helpful in those with the
MTHFR polymorphism, which was negative in the eight patients with CFD in whom we conducted gene testing. We continue to work to understand the biologic mechanism of our biochemical findings. It is clear that treatment with
l-methylfolate is not sufficient in these individuals, because of a deficit earlier in the folate metabolic pathway that produces bioactive metabolites. Furthermore, the administration of
l-methylfolate will falsely correct the CSF test to identify these individuals. While empirical treatment with folinic acid may seem appealing, this again is problematic with our current understanding. After folinic acid administration, definitive testing cannot be conducted, and the full effects of treatment with folinic acid in an individual with CFD occur over years. This time frame is due to neuronal turnover and growth in the setting of availability of active folate metabolites needed for appropriate white matter structure. We cannot determine whether the metabolite abnormalities are primary or secondary. However, our patients had no other overt clinical disease, and thus secondary deficiencies related to other systemic conditions are unlikely. Applications of next-generation sequencing technologies (i.e., whole exome or genome sequencing) on a focused, homogeneous patient population with treatment-refractory depression will provide further insight into genetic components of the metabolic findings in our patients. After further evaluation and characterization of a larger population of patients, animal models of biochemical abnormalities will be an important next step.
Although the majority of patients in this study were recruited by advertisement through a research registry, four participants were clinically referred, and the possibility of an ascertainment bias cannot be excluded. This was a relatively small sample of mostly Caucasian patients, and the treatment component of the study was an open trial. Continued treatment as usual resulted in medication changes during the course of the study, meaning that in three cases of CFD, there were concomitant medication changes in addition to folinic acid supplementation. Preliminary experience with treatment has been promising; however, long-term outcomes remain to be determined.
CSF testing is required to determine the presence of CFD and need for treatment. Review of the CSF metabolic profile directs the diagnosis and avoids missing other metabolic disorders that may be present.
l-Methylfolate (5-
l-methyltetrahydrofolate) is the treatment specific to methylenetetrahydrofolate deficiency (
32), but in the majority of cases, folinic acid is preferable because of its superior stability and lower costs when compared with
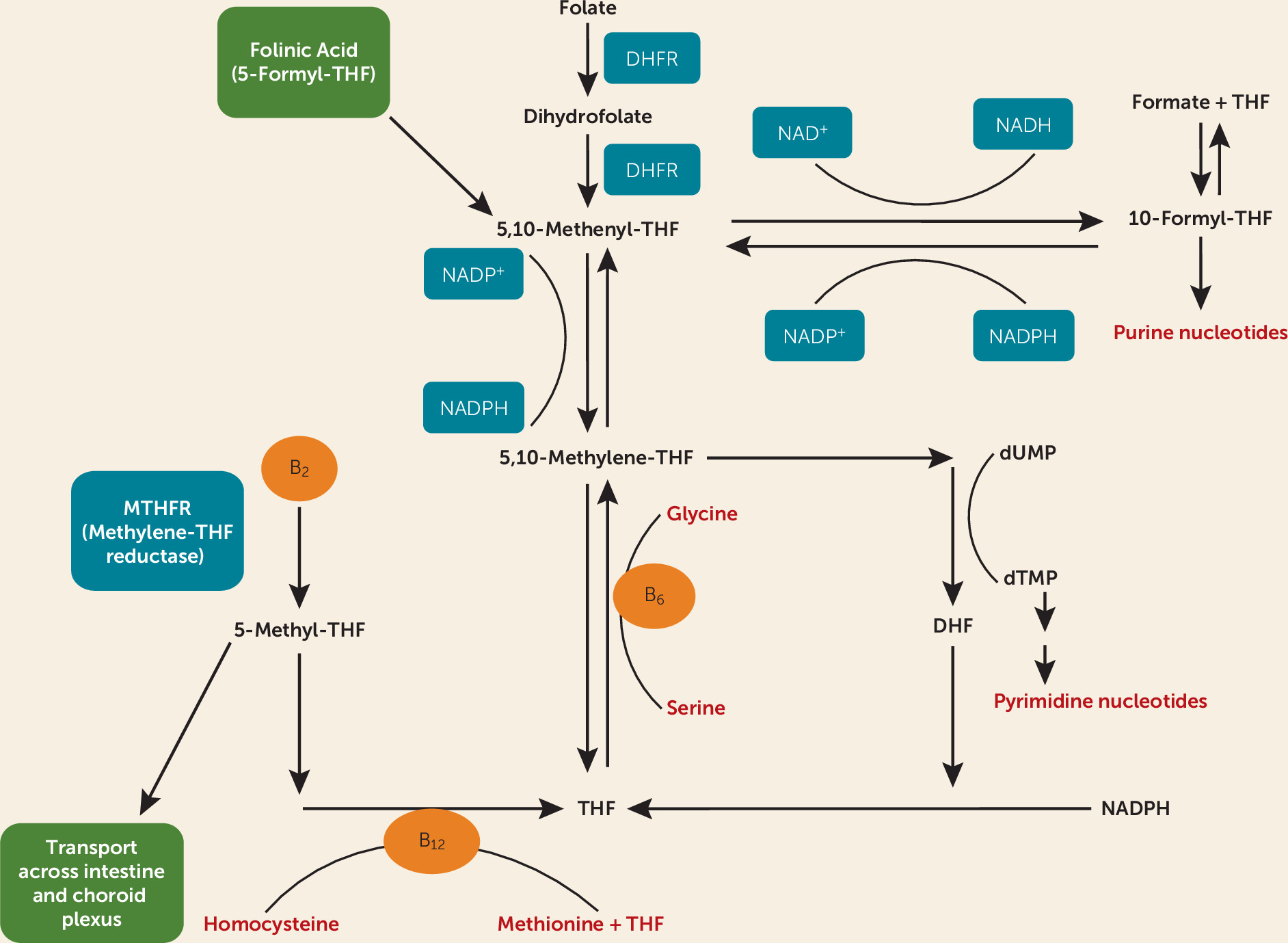
l-methylfolate. Dihydrofolate reductase converts folic acid to dihydrofolate and then to tetrahydrofolate (
Figure 2). Folinic acid (5-formyl-tetrahydrofolate) is converted to 5-,10-methenyltetrahydrofolate, then 5-,10-methylenetetrahydrofolate, and eventually to 5-MTHF. Folinic acid therefore allows the availability of active metabolites in the folate pathway in individuals with CFD that is not caused by methylenetetrahydrofolate deficiency. Furthermore, folinic acid has therapeutic effect in the setting of folate receptor autoantibodies, although there is controversy surrounding the assessment of these autoantibodies (
33).
Folinic acid should be given by prescription. Dosages of 1–2 mg/kg per day may be administered with prescription-strength folinic acid. Treatment with either folinic acid or
l-methylfolate will preclude testing after administration because they correct deficient CSF 5-MTHF levels. A clinical response should be expected in patients with treatment-refractory depression and low CSF 5-MTHF levels. Experience with more severe neurologic disorder associated with CFD (
34) indicates that the response may take several months, but the full effect of treatment may take 1–3 years. This may be related to neuronal growth and turnover in an environment in which folate metabolites are newly available. Folic acid is not recommended because of the potentially low activity of dihydrofolate reductase in CFD and because of its tight binding to folate receptor alpha, which results in reduced cerebral transport of 5-MTHF. Folinic acid treatment in our patients did have some side effects (facial flushing and initial anxiety). It is important to note that vitamin B
12 acts as a cofactor in this pathway. Blood folate and B
12 levels should be evaluated in all patients with treatment-refractory depression. The best option for patients with treatment-refractory depression for whom metabolic disorder is suspected is consultation with a biochemical geneticist.
Our findings indicate a need for further evaluation of the role of neurometabolic abnormalities in treatment-refractory depression. The remarkably high incidence of actionable abnormalities and some evidence of symptom improvement with treatment strongly support the need for larger studies. Historically, a diagnosis of a metabolic disorder has been considered in the context of a family history of a known disorder or symptoms that are exacerbated by significant physiologic stresses (e.g., fever, fasting, surgeries), especially with multisystem involvement (
35), neither of which was present in our patient population. Our findings, if replicated, suggest that neurometabolic disorders may contribute to treatment-refractory psychiatric disorders even without other systemic illness. An important question is whether early identification and treatment of an underlying metabolic abnormality early in the course of psychiatric illness could prevent long-term emotional and cognitive complications. If these findings are replicated, they suggest that the identification of new inborn errors of metabolism or secondary disorders of metabolism contributing to psychiatric illness may allow repurposing of currently approved orphan drugs.