Major depressive disorder is a highly prevalent psychiatric condition characterized by blunted reward processing and diminished positive affect (
1). Preclinical research has shown that phasic dopamine signaling, particularly in the striatum, constitutes an important neural mediator of reward-related behaviors, including reinforcement learning (
2,
3) and incentive motivation (
4). Functional MRI (fMRI) studies in humans have corroborated the central role of striatal function in reinforcement learning (
5) and reward processing (
6) and demonstrated that these striatal functions are disrupted in depression (
7,
8). Accordingly, reduced striatal dopamine functioning is believed to play a key role in the pathophysiology of depression, particularly in the context of impaired reward processing and reward learning (
9–
11). fMRI studies have further suggested that reward dysfunction in depression is related to disrupted corticostriatal functional connectivity (
12,
13), consistent with the notion that altered communication among dopamine-rich striatal regions and cortical regulatory systems is an important substrate of depression (
14).
Despite theories implicating striatal dopamine dysfunction in depression, it is unknown whether an acute manipulation thought to transiently increase dopamine signaling might normalize reward processing in depression. In healthy individuals, studies combining fMRI with acute pharmacologically induced dopaminergic enhancements have shown increased reward-related striatal responses and improved reward learning relative to placebo (
15–
17). For instance, acute administration of amisulpride (200 mg) improved healthy participants’ ability to select the better of two rewarding options, purportedly by enhancing reinforcement learning signals in the striatum and ventromedial prefrontal cortex (
15). However, no study to date has tested whether pharmacologically induced enhancement of dopaminergic transmission can improve reward learning or striatal activity and corticostriatal connectivity in response to reward in depression.
To address these important gaps in the literature, we conducted a double-blind randomized placebo-controlled study integrating neural and behavioral measures of reward processing in conjunction with a dopamine pharmacological challenge. To this end, 46 unmedicated depressed individuals and 43 healthy control subjects were randomly assigned to receive either placebo or a single low dose (50 mg) of the D
2/D
3 receptor antagonist amisulpride, which has a particularly high affinity for mesolimbic pathways and is believed to increase dopaminergic transmission by means of presynaptic D
2/D
3 autoreceptor blockade (
18,
19) (see also the Supplementary Methods section in the
data supplement that accompanies the online edition of this article). After administration of amisulpride or placebo, participants underwent fMRI scanning during a monetary incentive delay task involving anticipation and receipt of monetary rewards and penalties (
7). After the scan, participants completed a probabilistic selection task that separately measured the ability to learn from rewards or penalties (
20). We selected a 50-mg dose in light of previous reports that a (sustained) 50-mg dosage of amisulpride has antidepressant and antianhedonic effects in depressive disorders (
21,
22) and in order to avoid postsynaptic blockade (
23), with the goal of maximizing the likelihood of autoreceptor effects. We hypothesized that this pharmacological manipulation would be associated with increased striatal response to reward and improved reward learning, and that such effects would be largest among depressed individuals.
Method
Participants
Participants were recruited from the Boston metropolitan community. The depressed and control groups were matched for age, gender, ethnicity, and years of education (
Table 1). Inclusion criteria restricted recruitment to right-handed individuals 18–45 years of age with no contraindications to MRI, no lifetime substance dependence, no past-year substance abuse, and no serious medical conditions. For the depression group, participants had to have a diagnosis of major depressive disorder according to the Structured Clinical Interview for DSM-IV-TR Axis I Disorders (SCID) (
24). Exclusion criteria for the depressed group included use of any psychotropic medication in the past 2 weeks (6 weeks for fluoxetine, 6 months for dopaminergic drugs or antipsychotics) and a psychiatric history of other major axis I disorders. For the control group, inclusion criteria included medication-free status for at least 3 weeks, absence of current or past psychiatric illnesses (based on the SCID interview), and absence of first-degree familial psychiatric illness. Participants received $15/hour in compensation plus earnings in the fMRI task. All participants provided written informed consent to a protocol approved by Partners Human Research Committee.
Procedure
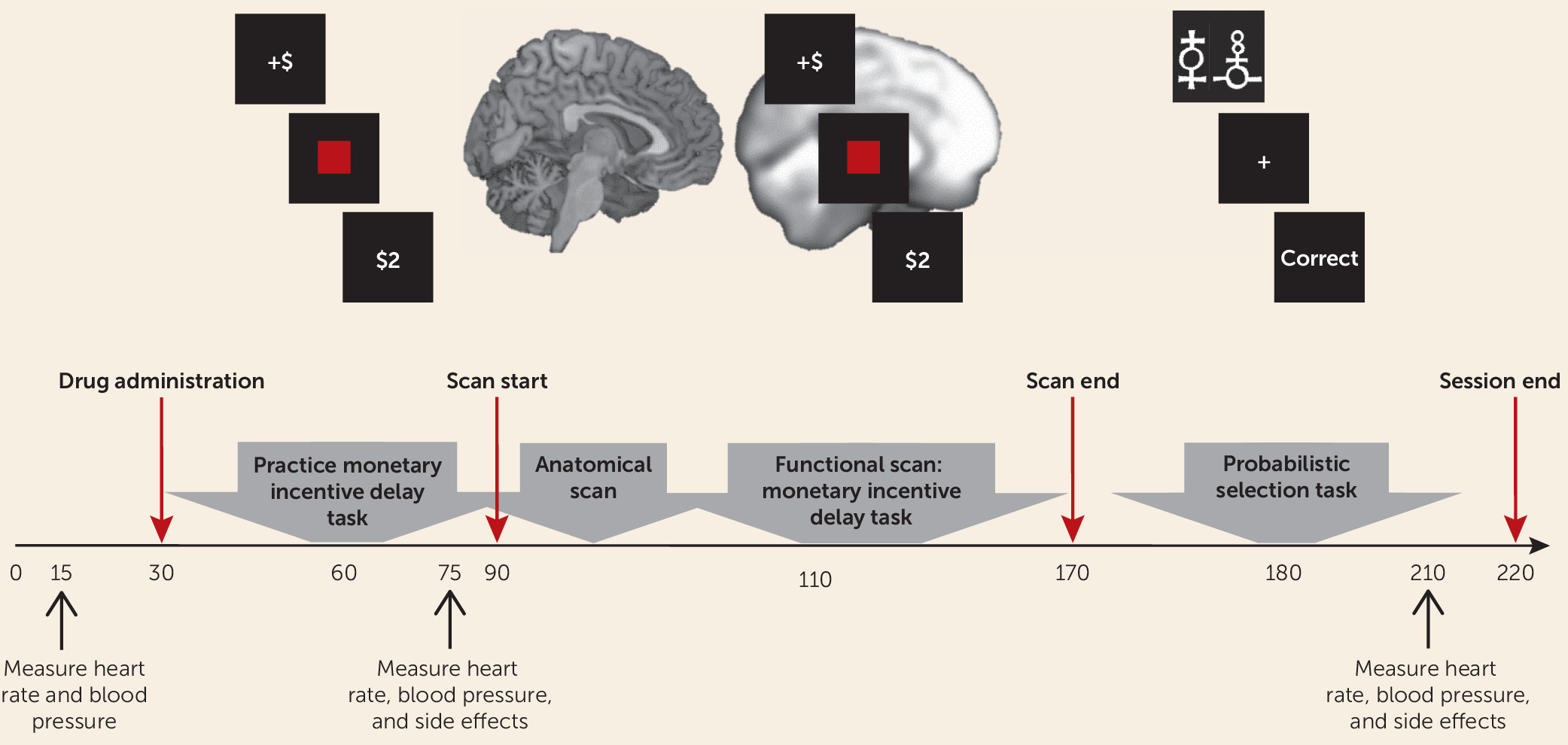
Participants first completed a clinical evaluation to determine eligibility (based on the SCID interview) and self-report measures of depression and anhedonia (
Table 1; see also the Supplementary Methods section in the
online data supplement). Eligible participants were invited to take part in the neuroimaging session, and those who participated were randomly assigned to receive amisulpride or placebo under double-blind conditions. Pharmacokinetic data indicate that plasma concentration of amisulpride has two peaks, approximately 1–1.5 hours and 2.5 hours after administration (
18,
19). Therefore, the study physician administered either amisulpride or placebo at the beginning of the neuroimaging session, and fMRI scanning of the monetary incentive delay task started 1 hour after amisulpride or placebo administration to coincide with the first plasma concentration peak. The probabilistic selection task was administered after scan completion, approximately 2.5 hours after amisulpride or placebo administration, to coincide with the second plasma concentration peak. Heart rate, blood pressure, and side effects were assessed by the study physician throughout the session (
Figure 1).
fMRI Task
The monetary incentive delay task involves anticipation and receipt of monetary rewards and penalties, which have been shown to elicit robust striatal response in healthy individuals (
25). Previous studies using this task have revealed reduced striatal activation and reduced corticostriatal functional connectivity in depressed compared with healthy adults during anticipation and receipt of monetary reward (
7,
26), making it well suited for the present study (see the Supplementary Methods section in the
data supplement).
Behavioral Task
A probabilistic selection task was used to probe learning from positive and negative feedback (
20). In the learning phase, participants repeatedly viewed three pairs of stimuli (AB, CD, and EF) and had to integrate feedback over several trials to learn which stimulus in each pair was rewarded most consistently. In the test phase, the most reliably rewarded (A) and penalized (B) stimuli were presented in conjunction with all other stimuli (e.g., AC, AD, AE, AF); participants’ ability to “choose A” or to “avoid B” were used as measures of reward or penalty learning, respectively (see the Supplementary Methods section in the
data supplement).
MRI Acquisition Parameters
The MRI acquisition parameters are described in the Supplementary Methods section of the data supplement.
fMRI Data Analysis
fMRI data were preprocessed using SPM12 (
http://www.fil.ion.ucl.ac.uk/spm/software/). Preprocessing included coregistration of functional and anatomical images, segmentation, nonlinear volume-based spatial normalization (using Montreal Neurological Institute [MNI] space), and spatial smoothing with a Gaussian filter (6 mm full width at half maximum).
Hemodynamic responses were modeled using a canonical hemodynamic response function that was convolved with the onset times of task regressors in order to compute a general linear model at the single-subject level. The general linear model included nine task-related regressors: three cues (reward, penalty, no incentive), the target, and five outcomes (win [reward outcome following reward cue], no win [no-change outcome following reward cue], loss [penalty outcome following penalty cue], no loss [no-change outcome following penalty cue], and no change [no-change outcome following no-incentive cue]). The general linear model also included high-pass temporal filtering (0.008 Hz), seven rigid-body movement parameters, nuisance regressors accounting for no-response trials, and outlier time points (see the Supplementary Methods section in the data supplement).
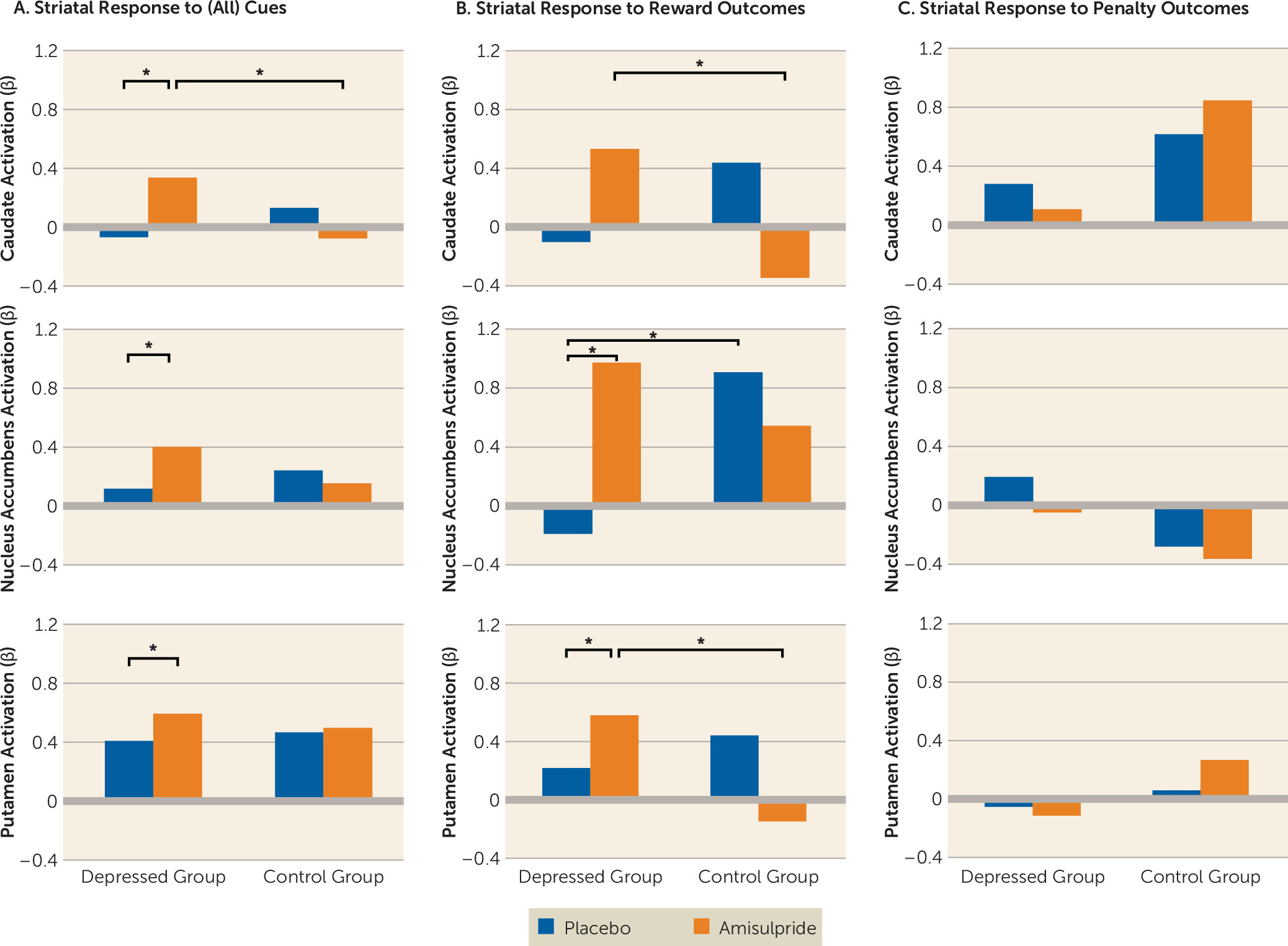
To test a priori hypotheses regarding striatal responses to reward (
7), we conducted a region-of-interest analysis in which activations (beta weights) were extracted from anatomical masks of the caudate, the nucleus accumbens, and the putamen for each participant and for each task regressor (relative to baseline). To avoid any biases, masks were defined using a manually segmented MNI-152 brain and implemented as overlays on the SPM12 canonical brain (see Figure S1 in the
data supplement; see also reference
27). Activations reported throughout the analyses were quantified by averaging beta weights from all voxels within a mask. Exploratory whole brain analyses were also conducted (see the Supplementary Methods and Results sections of the
data supplement).
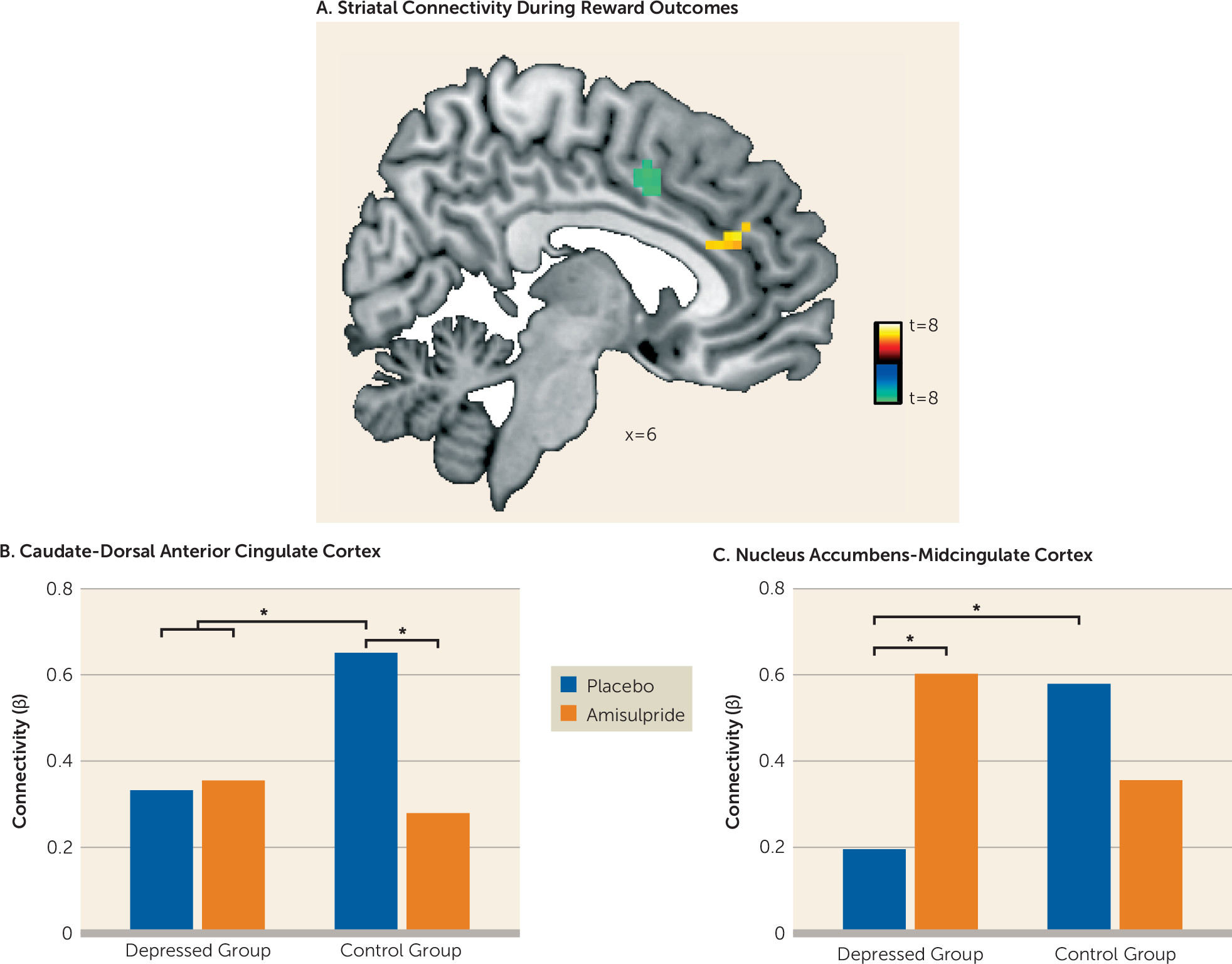
Psychophysiological interaction analyses were performed to examine the effects of reward and penalty outcomes on striatal functional connectivity. Because hemispheric effects on task activation were nonsignificant, striatal masks were collapsed across hemispheres, yielding three bilateral seeds (caudate, nucleus accumbens, putamen). Analyses retained the subject-level general linear models described above, adding regressors corresponding to the seed time course and the interaction of the seed time course with the task condition of interest (separately for reward and penalty outcome). Single-subject connectivity maps for the interaction between each seed time course and the regressor of interest were entered into second-level whole brain random-effects analysis. Effects were thresholded at a peak p value of <0.001, whole brain family-wise error corrected to p<0.05 at the cluster level.
Statistical Analysis
The methods for statistical analysis are detailed in the Supplementary Methods section of the online data supplement.
Discussion
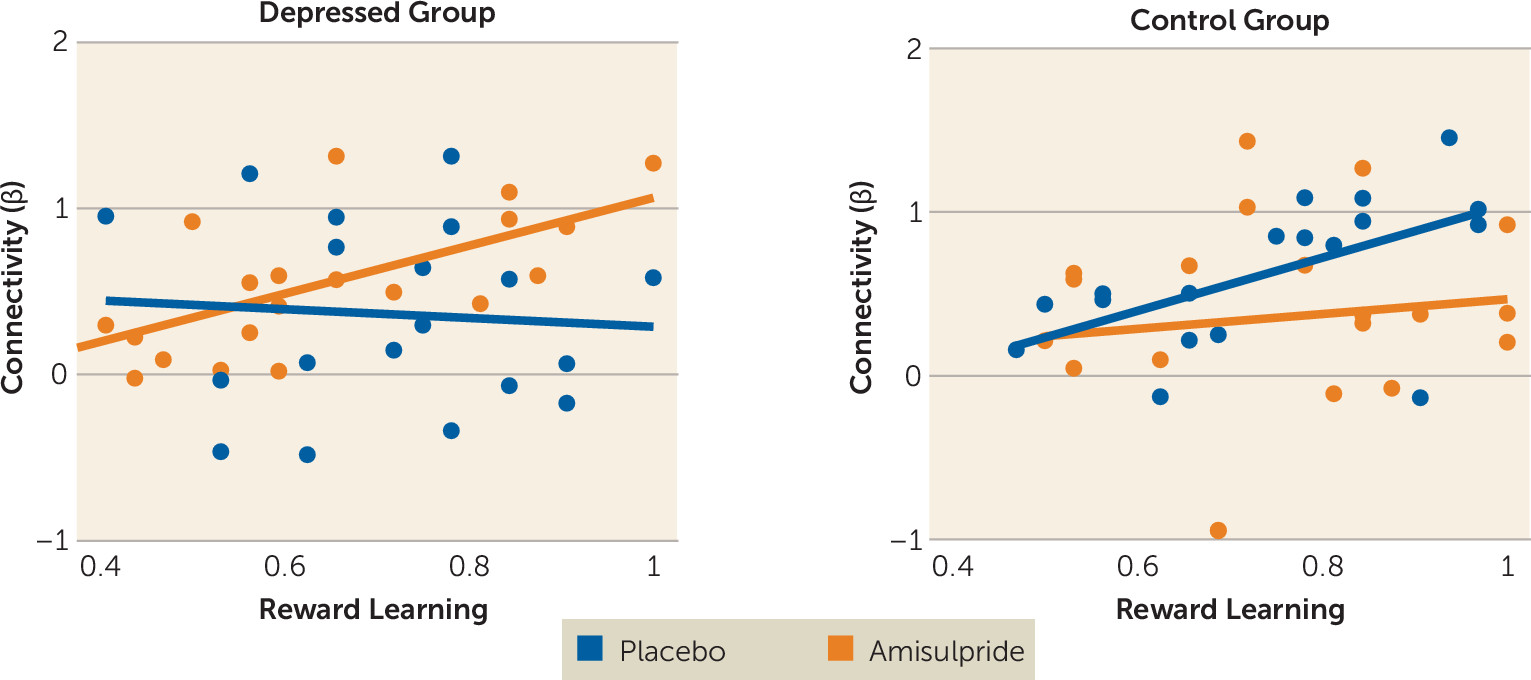
Major depression is a debilitating psychiatric disorder characterized by high rates of relapse and recurrence. Discovering treatment tools that target putative mechanisms of illness in depression—such as blunted response to reward—is therefore a key clinical priority. Findings from this proof-of-mechanism study suggest that an acute pharmacological challenge transiently increased striatal response to reward among adults with major depressive disorder, putatively via enhancement of dopaminergic transmission owing to autoreceptor blockade. Specifically, depressed participants receiving amisulpride exhibited increased striatal activity in response to cues, and increased striatal activity and corticostriatal functional connectivity in response to reward outcomes. Furthermore, stronger corticostriatal functional connectivity between the nucleus accumbens and the midcingulate cortex in depressed participants who received amisulpride was associated with better reward learning performance, a pattern similar to that observed in healthy control subjects receiving placebo. Together, these results provide converging evidence for abnormalities in neural reward systems in depression and highlight the potential of targeted pharmacological treatments to normalize reward processing in depression.
Extensive preclinical research has emphasized the key role of striatal dopamine signaling in mediating reward-related behaviors (
2–
4) and has postulated links between reduced striatal dopamine function and blunted reward processing and reinforcement learning in depression (
9,
10). Interestingly, previous research indicates that dopamine differentially mediates anticipatory and consummatory phases of reward processing (
28) and thus may uniquely affect their putative dysfunction in anhedonia and depression (
29). In support of this idea, we observed that acute administration of amisulpride enhanced striatal response to cues regardless of valence (e.g., signaling potential rewards, penalties, or null outcomes), yet in response to outcomes, striatal enhancement was selective to reward.
In addition to increasing striatal activity in response to rewards, enhancement of dopamine signaling in depressed individuals was also associated with amplified functional connectivity between the striatum and areas of the midcingulate cortex. This finding is consistent with a model in which abnormal coordinated activity among large-scale brain circuits, including corticostriatal pathways, is central to the pathophysiology of depression (
30,
31). Critically, those depressed individuals who exhibited the strongest nucleus accumbens–midcingulate cortex connectivity in response to rewards after amisulpride administration also exhibited better reward learning in an independent behavioral task, and this pattern was not found among depressed individuals who received placebo. Of relevance to the present findings, increased functional connectivity has been observed between midcingulate and striatal regions (and the insula) during learning (
32), supporting the importance of this corticostriatal subcircuit in dopamine-mediated functioning. Coordination between dopamine-rich areas of the striatum and midline regions involved in processing behavioral salience may therefore be an important dimension of healthy reinforcement learning, and dopamine enhancement may help to regulate this functional circuit in depression. In fact, given preclinical evidence that amisulpride has a particularly high affinity for mesolimbic pathways (
18,
19), one may speculate that amisulpride may enhance striatal function by affecting regulatory mechanisms beyond the striatum, and in particular in regions of the mesocorticolimbic pathway that communicate with the striatum via dopaminergic signaling to enable reward motivation and reinforcement learning (
29). Thus, while in the present study we investigated the effects of amisulpride on striatal functioning, other brain systems that have exhibited abnormal activity or functional connectivity in depression (e.g., prefrontal cortex) may be important targets of dopamine manipulation.
Several additional questions remain open for future investigation. First, evidence from preclinical studies linking reinforcement learning and motivation with phasic dopamine signaling in the striatum suggests that amisulpride enhancement of reward processing in depressed individuals most likely occurs via increased phasic dopamine signaling (
2–
4). Nevertheless, the mechanisms by which amisulpride may act to enhance striatal response to reward are complex and may involve modifications of phasic and tonic levels of dopamine, as well as of additional neurotransmitters (
33,
34). Additional research, especially in humans, investigating the effects of amisulpride on tonic and phasic dopamine release is needed. A second area for future investigation is motivated by differences between our findings and the results of previous investigations in which dopaminergic manipulation in healthy individuals resulted in better reward learning and increased striatal activity. The modest amisulpride dose used in the present study (50 mg as opposed to 200 mg and 400 mg in past studies [
15,
35]) may have contributed to these discrepancies. We selected a 50-mg dose based on animal work showing that low doses of amisulpride potentiate striatal dopamine release, have strong hedonic effects, and increase the incentive value of environmental cues (
18,
19). In humans, a 50-mg dose of amisulpride has been associated with reduced blockade of postsynaptic D
2/D
3 receptor in comparison to higher doses of 200–400 mg (
23), increasing the likelihood of presynaptic effects. Perhaps more importantly, (sustained) 50-mg amisulpride dosing has been shown to have antidepressant and antianhedonic effects in depressive disorders (
21,
22), suggesting that the present pharmacological manipulation may preferentially benefit depressed individuals as compared with healthy subjects. Nevertheless, while the pharmacological manipulation enhanced striatal function in depressed individuals, it had no such effect on behavior (i.e., reward learning). One potential reason for this could relate to the fact that we administered only a single dose of the drug. Thus, while the drug may have an immediate effect on neural function, modifying behavior may require longer and more chronic exposure. In support of this idea, antidepressant effects of amisulpride among depressed individuals have been observed after sustained (but not acute) administration (
21,
22).
In conclusion, in depressed individuals, but not healthy subjects, acute pharmacological challenge transiently increased striatal activity and corticostriatal functional connectivity in response to rewards, putatively via enhancement of dopaminergic transmission. These findings suggest that an acute pharmacological manipulation believed to increase dopamine transmission may help normalize reward processing in depressed individuals through the enhancement of key corticostriatal mechanisms.