Developmental Delay, Treatment-Resistant Psychosis, and Early-Onset Dementia in a Man With 22q11 Deletion Syndrome and Huntington’s Disease
This case provides an example of genetic conditions associated with “copy number variation,” where portions of the genome are either excessively repeated as “copies” (i.e., duplications) or where normal “copies” are absent (i.e., deletions). The early-life developmental abnormalities in this patient were associated with what?
Past Medical History
Family History
Investigations
Neurological Examination
Functional status.
Physical examination.
Mental status examination.
Neurological examination.
Genetic Analyses
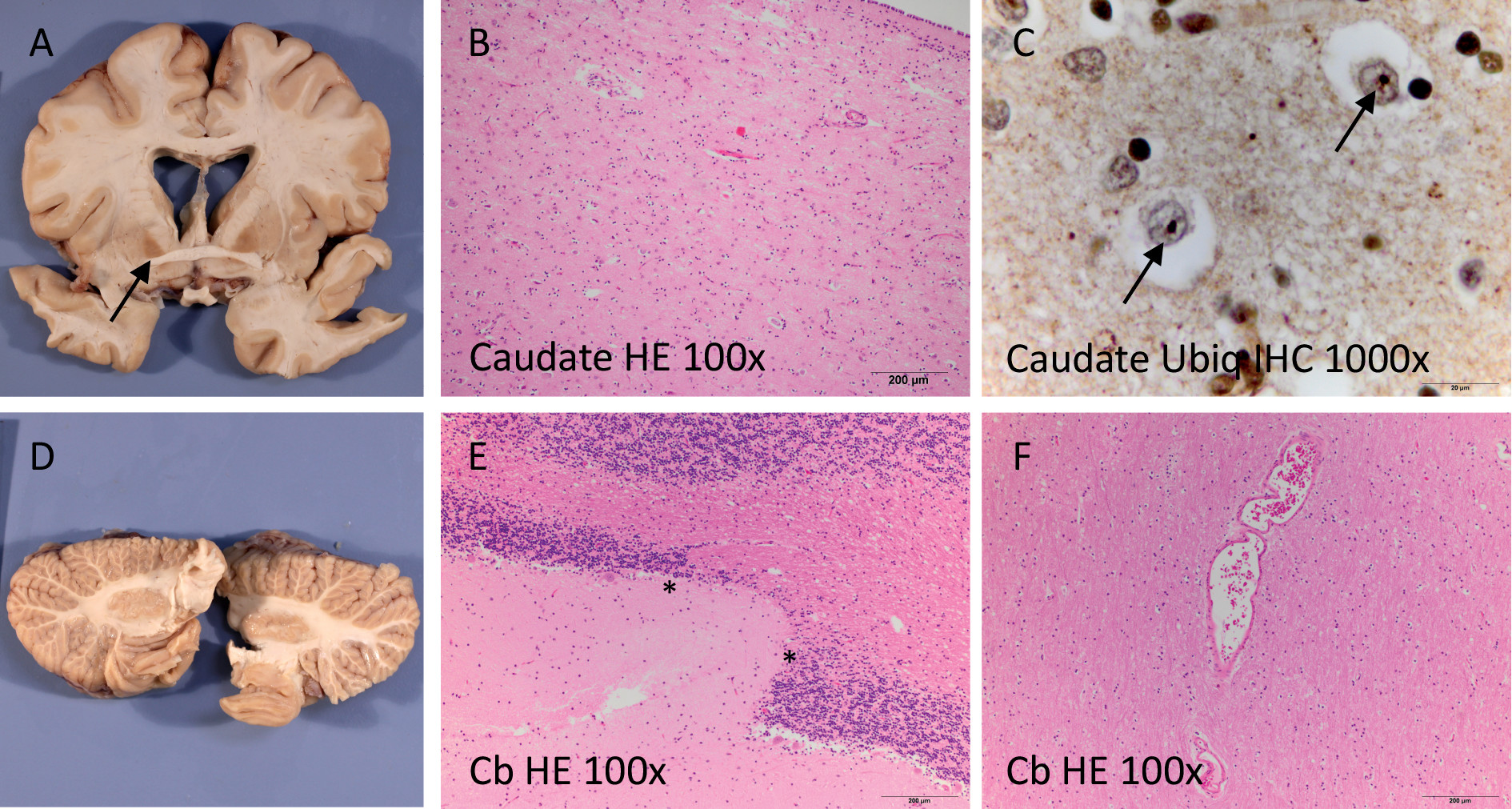
Death and Pathological Examination
| Age | Life Event | Clinical Notes |
|---|---|---|
| 0 | Birth | Breech birth, hernia, cleft palate, protuberant ears |
| 3 months | Surgery | Two herniorrhaphies |
| 6 months | Adopted | |
| 4 years | Delayed milestones | Preschool staff did not view him as ready for kindergarten compared with peers |
| Surgery | Surgery to correct cleft palate | |
| Grades K–5 | Elementary school | Learning disability; “warm, sociable” |
| Grades 6–8 | Middle school | Special education classes. IQ measured at 80, dyslexia, auditory processing problems. Surgery to correct protuberant ears |
| 14–19 years | High school | Graduated 2 years behind grade level; angry, possible substance misuse |
| 20 years | College | Brief college attendance. Few friends, abused by peers, possible substance misuse |
| 21 years | Psychotic episodes resulting in two admissions to community psychiatric hospital (14 days each) and day treatment | Delusions of strangers surrounding house with intent to do physical harm, auditory hallucinations, hitting mother, throwing objects. GAF score, 20–25. Medications: perphenazine, lithium |
| 21–22 years | Three inpatient admissions in university psychiatry unit for psychosis (1 month each) | Intense paranoia, auditory hallucinations, aggressive threats, suicidal ideation. GAF score, 10–20. Brain MRI was unremarkable. Thrombocytopenia. Medications: perphenazine, olanzapine, clozapine |
| 22–27 years | Long-term state inpatient hospitalization | Admission GAF score, 20, due to severe paranoia and auditory hallucinations. Poor initial response to clozapine, but good response with adequate dose and duration. Obesity. No extrapyramidal symptoms and normal gait noted on annual medical/neurological examination. Thrombocytopenia. Medications: clozapine, risperidone, fluoxetine |
| 27–29 years | 24-hour supervised community living program | Few delusions or auditory hallucinations; the patient was actively involved in discharge processing; vocational training recommended as part of therapy program. GAF score, ∼60. Medications: clozapine, ziprasidone |
| 29 years | Acute psychotic episode, medication abruptly stopped; 7-month inpatient admission | Extensive bruising, fetal position, incontinent, incoherent, randomly swinging fists. Thrombocytopenia. Medications: clozapine |
| 29–41 years | 24-hour supervised community living program | Initially relatively functional; able to do most self-care, worked in sheltered environment, participated in group outings, could play video games. Decline in function beginning in early 30s. By his late 30s, marked cognitive impairment, impaired gait, incontinence, incoherence, randomly swinging fists, language limited to grunting, required 24-hour one-to-one care. GAF score, 10–20. Loss of speech prominent at age 37. Thrombocytopenia (workup unrevealing). Medications: clozapine, lorazepam (for agitation), p.r.n. temazepam, melatonin (for sleep) |
| 37 years | Psychiatric consultation (second opinion) | Prompted by decline in function compared with early 30s: decline in activities of daily living (had done most self-care independently, e.g., personal hygiene, making meals); worsening performance in sheltered work environment; lessened interest in activities that had previously been of interest. Staff had initiated plan to try to correct lessened verbal output (e.g., from lengthy responses to queries to very brief responses). Examination: halting/listing gait; inconsistent responses to questions; bizarre behavior (e.g., standing in center of room in “almost catatonic state”). Occasionally incontinent of stool and urine. Brain MRI: poor quality due to motion, read as unremarkable. Thrombocytopenia, hypocalcemia |
| 39 years | Admission to inpatient neurology unit at a major academic center | Cognitive decline now severe. No expressive language, minimal or no comprehension. Evaluated for frontotemporal dementia. Brain MRI without contrast: poor quality due to motion, and terminated prematurely because the patient was unable to cooperate, but read as no gross intracranial mass or disproportionate atrophy. No movement disorder noted. Discharged back to community living program |
| 41 years | Enrolled in precision medicine study | Review of all records, genetic testing, examination by behavioral neurologist |
| 41 years | Death | Autopsy (cause of death, lobar pneumonia), neuropathology |
Perspective From Mr. E’s Mother
Recommendations and Conclusions
C. A deletion on chromosome 22.
Acknowledgments
References
Information & Authors
Information
Published In


History
Keywords
Authors
Author Contributions
Funding Information
Metrics & Citations
Metrics
Citations
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
For more information or tips please see 'Downloading to a citation manager' in the Help menu.
View Options
View options
PDF/EPUB
View PDF/EPUBLogin options
Already a subscriber? Access your subscription through your login credentials or your institution for full access to this article.
Personal login Institutional Login Open Athens loginNot a subscriber?
PsychiatryOnline subscription options offer access to the DSM-5-TR® library, books, journals, CME, and patient resources. This all-in-one virtual library provides psychiatrists and mental health professionals with key resources for diagnosis, treatment, research, and professional development.
Need more help? PsychiatryOnline Customer Service may be reached by emailing [email protected] or by calling 800-368-5777 (in the U.S.) or 703-907-7322 (outside the U.S.).