Major depressive disorder (hereafter referred to as “depression”) is moderately heritable (twin-based heritability, ∼37%) (
1), but its genetic architecture is complex, and identifying specific polymorphisms underlying depression susceptibility has been challenging. With the ability to genotype particular genetic variants and optimism about the potential public health impact of identifying reliable biomarkers for depression (
2), early research focused on the effects of specific candidate polymorphisms in genes hypothesized to underlie depression liability. These genes were chosen on the basis of hypotheses regarding the biological underpinnings of depression. The 5-HTTLPR variable number tandem repeat (VNTR) polymorphism in the promoter region of the serotonin transporter gene
SLC6A4, the most commonly studied polymorphism in relation to depression (
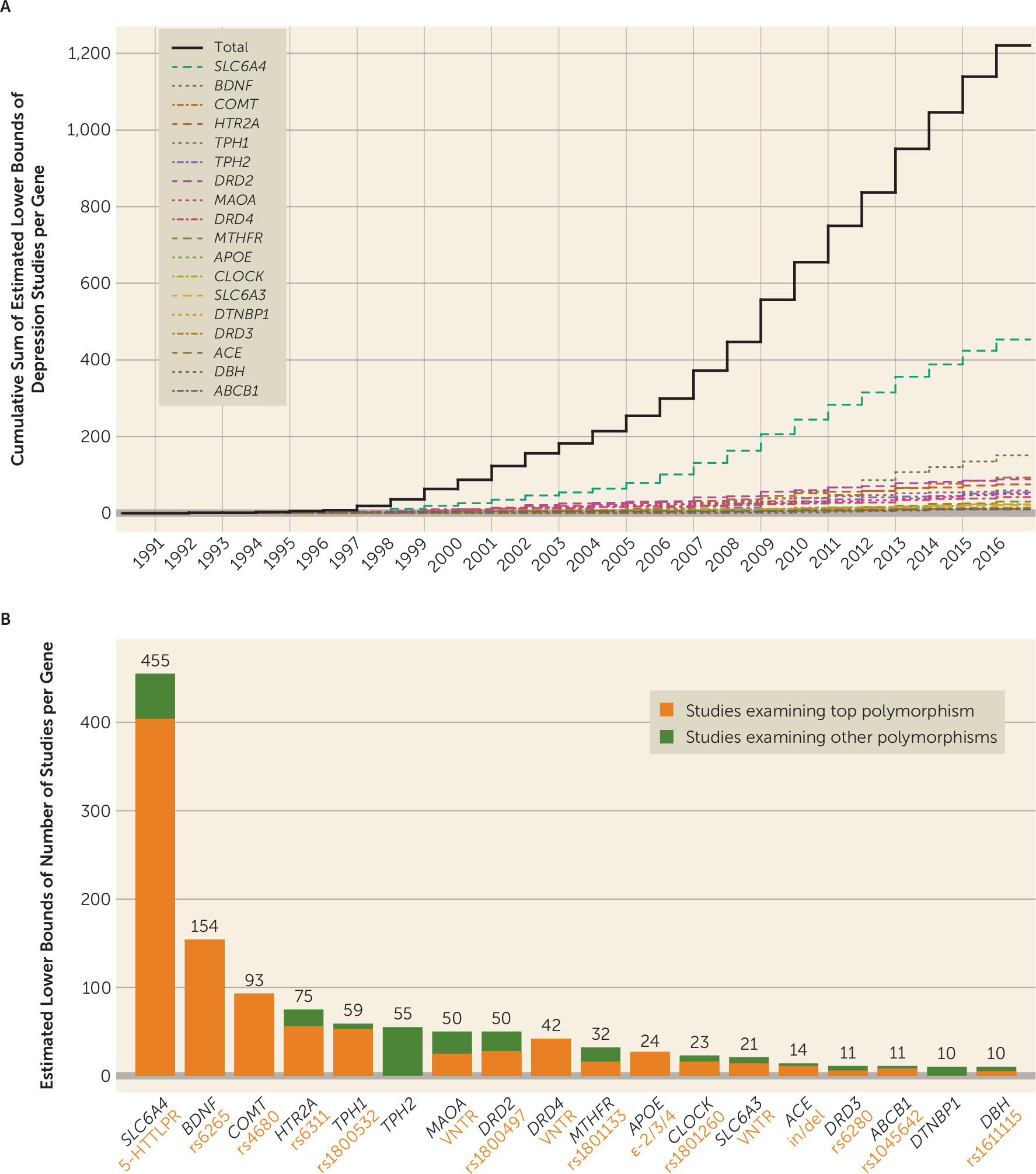
Figure 1; see also Table S1.1 in the
online supplement), serves as a prototypical example: Given the theorized importance of the serotonergic system in the etiology of depression, a logical target for early association studies was a common, large (and hence relatively easy to genotype), and potentially functional repeat polymorphism in a serotonergic gene (
3–
5). Early investigations, although by necessity focused on a small number of variants (low-cost genome-wide arrays were not yet available), reported promising positive associations. However, replication attempts produced inconsistent results (
6–
8).
To critics of candidate gene findings, replication failures suggested that the initial findings were artifactual (
9–
11). However, at least two alternative explanations could account for the inability to replicate early findings and the inconsistent results across studies. First, in the early 2000s, Caspi et al. (
12) posited that previous inconsistencies might reflect the effects of candidate polymorphisms that were dependent on environment exposures (gene-by-environment interaction [G×E] effects). In what would become one of the most highly cited (>8,000 citations as of July 2018) and influential papers in psychiatric genetics, Caspi et al. (
13) reported that the impact of the 5-HTTLPR repeat polymorphism in
SLC6A4 on depression was moderated by exposure to stressful life events, such that the positive association between stressful life events and depression was stronger in individuals carrying the “short” allele. This early work led many researchers to shift their attention to G×E hypotheses, focusing on the same polymorphisms first investigated for main effects (
8). Second, in an alternative but complementary line of reasoning, other researchers suggested that polymorphisms in the same candidate genes, other than those studied previously, were likely to explain depression risk, given the genes’ putative biological relevance (
14). These lines of inquiry are well represented in the literature of the past 25 years. Thousands of investigations of depression or depression endophenotypes have examined 1) the direct effects of the most studied polymorphisms within candidate genes, 2) the moderation of their effects by environmental stressors, or 3) the effects of alternative polymorphisms within the same candidate genes. The popularity of these lines of inquiry has not diminished over time (
Figure 1; see also Figures S1.4 and S1.5 of the
online supplement), and many studies have reported statistically significant associations.
Perhaps surprisingly given the continued interest in studying these historical depression candidate genes and the large number of associations documented in the candidate gene literature, many researchers have expressed skepticism about the validity of such findings (
11,
15–
17). There are several reasons for this. First, genome-wide association studies (GWASs), which agnostically examine associations at millions of common single-nucleotide polymorphisms (SNPs) across the genome in large samples, have consistently found that individual SNPs exert small effects on genetically complex traits such as depression (
18–
20). For example, in the most recent GWAS of depression, which utilized a sample of 135,458 case subjects and 344,901 control subjects, the strongest individual signal detected (rs12552; odds ratio=1.044, p=6.07×10
−19) would require a sample of approximately 34,100 individuals to be detected with 80% power at an alpha level of 0.05, assuming a balanced case-control design (
18). In contrast, the median study sample size in a review of 103 candidate G×E studies published between 2000 and 2009 was 345, with 65% of studies reporting positive results (
15). Thus, given the small sample sizes typically employed, candidate gene research has likely been severely underpowered (
21,
22). This, in turn, may suggest that the false discovery rate for the many positive reports in the candidate gene literature is high. Consistent with this possibility, targeted, well-powered genetic association studies of depression and other psychiatric phenotypes in large samples have not supported candidate gene hypotheses (
18,
23–
27). For example, a preregistered collaborative meta-analysis of the interaction of stressful life events and 5-HTTLPR genotype in a sample of 38,802 individuals failed to support the original finding of Caspi et al. (
28), although we note that this variant and several other candidate VNTRs have not previously been examined in a GWAS context (
29,
30). The absence of previous large-sample investigations of VNTR hypotheses is noteworthy, as VNTRs comprise several of the earliest candidate polymorphisms to be examined in the context of behavioral research; concerns about variability in VNTR genotyping procedures and analytic methods over time have further complicated the interpretation of the literature (
31). Additionally, a number of researchers have suggested that incorrect analytic methods and inadequate control for population stratification characterize the majority of published candidate gene studies (
21,
32–
34), and other researchers have questioned the clinical utility of focusing on individual polymorphisms or polymorphism-by-environment interactions (
35). Finally, there is evidence of systematic publication bias in the candidate gene literature; in the aforementioned review of all candidate G×E studies published between 2000 and 2009, 96% percent of novel findings were significant, compared with only 27% of replication attempts, and replication attempts reporting null findings had larger sample sizes than those presenting positive findings (
15). In response to such skepticism, candidate gene proponents have argued that lack of replication of candidate gene associations in large-sample studies may reflect poor or limited phenotyping (
36–
38), exclusion of non-SNP polymorphisms such as VNTRs (
14,
30), the “multiple-testing burden” associated with genome-wide scans (
36), and failure to account for environmental moderators (
36,
37,
39).
The present study is the most comprehensive and well-powered investigation of historical candidate polymorphism and candidate gene hypotheses in depression to date. We focus on three lines of inquiry concerning how historical candidate genes may affect depression liability: 1) main effects of the most commonly studied candidate polymorphisms, 2) moderation of the effects of these polymorphisms by environmental exposures, and 3) main effects of common SNPs across each of the candidate genes.
We first empirically identified 18 commonly studied candidate genes represented in at least 10 peer-reviewed depression-focused journal articles between 1991 and 2016 from the body of publications indexed in PubMed. Within these candidate genes, we identified the most commonly studied polymorphisms, as well as their canonical risk alleles, at which point our primary analysis plan was preregistered. Using multiple large samples (Ns ranging from 62,138 to 443,264 across subsamples; total N=621,214 individuals), we examined multiple measures of depression (e.g., lifetime diagnostic status, symptom severity among individuals reporting mood disturbances, lifetime number of depressive episodes) (
Table 1), employing multiple statistical frameworks (e.g., main effects of polymorphisms and genes, interaction effects on both the additive and multiplicative scales) and, in G×E analyses, considering multiple indices of environmental exposure (e.g., traumatic events in childhood or adulthood). Previous large-sample studies of depression have largely focused on genetic main effects on depression diagnosis in the context of SNP data across the genome. In contrast, we examined several alternative depression phenotypes, analyzed both main effects and interactions with multiple potential moderators, included the most studied polymorphisms, including VNTRs (
Figure 1), and employed a liberal significance threshold. We also quantified the extent to which phenotypic measurement error may have biased our results. The unifying question underlying this “multiverse” analytic approach (
44) was the following: Do the large data sets of the whole-genome-data era support any previous depression candidate gene hypotheses?
Methods
Identification of Genes and Polymorphisms
Using the Biopython bioinformatics package (
45), we identified 18 candidate genes studied for their associations with depression phenotypes at least 10 times from within the body of peer-reviewed biomedical literature indexed in PubMed. We used regular expressions to find articles potentially corresponding to each gene and hand-verified the number of correctly classified articles for each gene in order to estimate hypergeometric confidence intervals for the true number of correctly classified studies (for additional details, see section S1 of the
online supplement). We identified single polymorphisms comprising a large proportion of study foci for 16 of the 18 candidate genes.
Figure 1 lists the most studied candidate genes and polymorphisms within them, as well as probabilistic estimates of the minimum number of times each has been studied with respect to depression and the number of studies per gene per year (confidence intervals are presented in Table S1.1 in the
online supplement).
Samples
UK Biobank samples.
A large portion of the data used in our analysis was collected by the UK Biobank, a population sample of 502,682 individuals collected at 22 centers across the United Kingdom between 2006 and 2010 (
46). Within this group, we analyzed several depression phenotypes and moderators among 177,950 unrelated (pairwise genome-wide relatedness, <0.05) European-ancestry individuals for whom relevant depression measures were collected. We analyzed two partially overlapping subsets of these individuals: 91,121 individuals for whom selected items from the initial touchscreen interview were available and 115,458 individuals who completed a series of online mental health questionnaires, 62,138 of whom endorsed a 2-week period characterized by anhedonia or depressed mood at some point during their lives. DNA was extracted from whole blood and genotyped using the Affymetrix UK Biobank Axiom array or the Affymetrix UK BiLEVE Axiom array and imputed to the Haplotype Reference Consortium by the UK Biobank (
47). Further details on genotyping and sampling procedures are available online (
48) and in section S2 of the
online supplement. Because VNTRs were not genotyped in the UK Biobank data set, we used two independent whole-genome SNP data sets (the Family Transition Project [
49] and the Genetics of Antisocial Drug Dependence [
50,
51]) that also measured these repeat polymorphisms as reference panels in order to impute highly studied VNTRs within
DRD4,
MAOA,
SLC6A3, and
SLC6A4 in the UK Biobank. The estimated out-of-sample imputed genotype match rates were ≥0.919 for all four VNTRs (mean R
2=0.868; details are provided in reference
29).
Psychiatric Genomics Consortium sample.
To investigate candidate gene polymorphism main effect hypotheses, we also used data from the most recent GWAS on depression conducted by the Major Depressive Disorder Working Group of the Psychiatric Genomics Consortium (PGC), which is described in detail in Wray et al. (
18). Lack of access to raw genotypes for a large number of the PGC cohorts precluded imputation of VNTRs in the PGC sample. To minimize sample overlap with the UK Biobank, U.K.-based cohorts were excluded from the PGC data set, resulting in GWAS summary statistics for a total of 443,264 individuals (120,201 case subjects and 323,063 control subjects) (for further details, see section S2 of the
online supplement).
Phenotypes
Table 1 describes all phenotypes examined in the present investigation, and additional information is provided in section S3 of the
online supplement. Correlations between depression outcomes and Cohen’s kappa estimates for diagnosis phenotypes are presented in Tables S3.1 and S3.2 in the
online supplement. Marker-based heritabilities of, and genetic correlations between, depression outcomes were estimated via linkage disequilibrium (LD) score regression (
52) and are presented in Tables S3.3 and S3.4 and Figure S3.3 in the
online supplement (for further details, see section S4.4 of the
online supplement).
Analyses
All analyses were preregistered through the Open Science Framework and are available at
https://osf.io/akgvz/. Statistical models are described in detail in section S4 of the
online supplement, and departures from the preregistered analyses are documented in section S5.
Polymorphism-wise analyses.
We analyzed associations between outcomes and each of the top 16 candidate polymorphisms using a generalized linear model framework (link functions are listed in Table S4.1 in the
online supplement). For two of the genes,
TPH2 and
DTNBP1, no particular polymorphism was investigated in a preponderance of studies (see Figures S1.2 and S1.3 in the
online supplement), so these genes were not included in the polymorphism-wise analyses. Covariates included genotyping batch, testing center, sex, age, age squared, and the first 10 European-ancestry principal components. Sixteen polymorphism-by-environment effects were tested on both the additive and multiplicative scales for each of the 16 polymorphisms; each model tested is listed in Table S4.1 in the
online supplement. For interaction tests, we included all covariate-by-polymorphism and covariate-by-moderator terms to control for the potential confounding influences of covariates on the interaction (
53). We also tested interaction models that controlled only for covariate main effects, which is insufficient but common in the candidate gene literature (
33). Across all outcomes, we employed a preregistered significance threshold of alpha
poly=0.05/16=3.13×10
−3, corresponding to a Bonferroni correction across the top 16 candidate polymorphisms. This threshold is liberal because it does not account for the multiple ways each polymorphism was analyzed or the multiple outcomes it was assessed with respect to. Further details are provided in section S4.1 of the
online supplement.
Gene-wise and gene-set analyses.
We used the National Center for Biotechnology Information (NCBI) Build 37 gene locations to annotate SNPs to genes, allowing SNPs within a 25-kb window of the gene start and end points to be mapped to each gene. We used MAGMA, version 1.05b (
54), to perform gene-wise and gene-set analyses for the top 18 candidate genes separately in the UK Biobank and PGC data sets. Gene-wise tests summarize the degree of association between a phenotype and polymorphisms within a given gene; in contrast, gene-set tests examine the association between a phenotype and a set of genes rather than individual genes.
We conducted gene-wise association analyses for each gene and outcome using the MAGMA default gene-level association statistic (sum −log p-based statistics and principal components regression, for tests based on summary statistics and individual-level genotypes, respectively) and using a liberal significance threshold of alpha
gene=0.05/18=2.78×10
−3 to correct for multiple tests across the 18 candidate genes. We used summary statistics from the PGC2 depression GWAS (
18) (excluding UK-based cohorts) as input for the PGC analyses, whereas individual-level genotypes were available for the UK Biobank. The gene-level association statistics were in turn used to perform “competitive” gene-set tests that compared enrichment of depression phenotype–associated loci between our set of 18 candidate genes and all other genes not in the gene set, controlling for potentially confounding gene characteristics. Further analyses, which compared the 18 candidate genes to negative control sets of genes involved in type 2 diabetes, height, or synaptic processes, are described in section S4.2 of the
online supplement, and results are reported in section S11.
Results
Polymorphism-Level Analyses
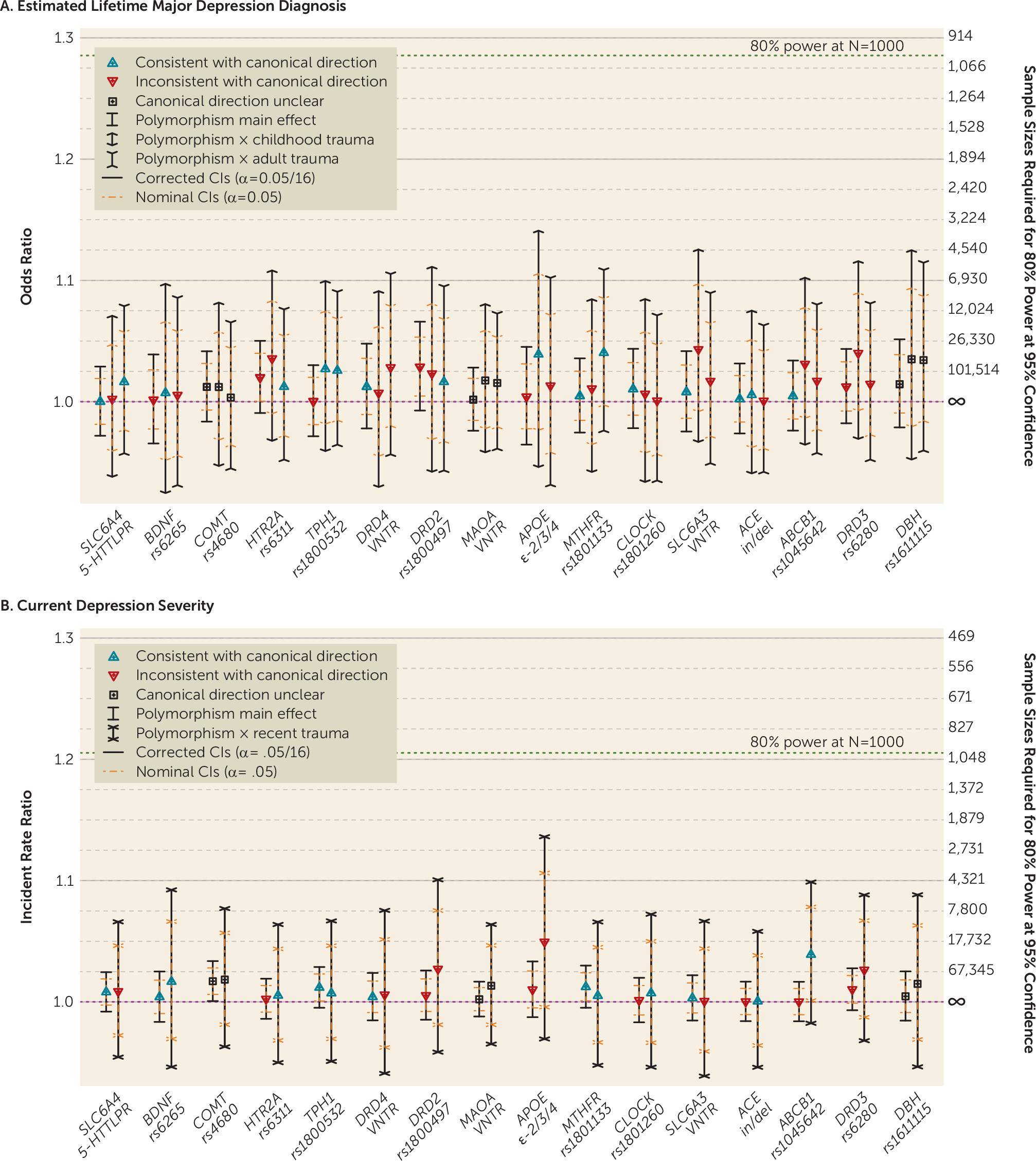
Table 2 lists the most significant result for each of the most-studied candidate gene polymorphisms for the main effect across the eight outcomes investigated (eight main effect tests per polymorphism) and the interaction effect across five moderators measured in the UK Biobank (32 interaction tests per polymorphism [see section S4.1 of the
online supplement]). Given the number of tests conducted, there was little evidence that any effect was larger than what would be expected by chance under the null hypothesis. Only for
COMT rs4680 on current depression severity was there was evidence of a small main effect that surpassed our liberal threshold of significance, such that the incident rate of current depression severity scores decreased by a factor of 0.983 per copy of the G allele (odds ratio 95% CI=0.967–0.999; p=0.002) (
Figure 2). Detecting an effect of this size at an alpha level of 0.05 with 80% power would require a sample of over 100,000 individuals (see section S4.3 of the
online supplement). Similarly, across all polymorphisms, outcomes, and exposures, on both the additive and multiplicative scales, no polymorphism-by-exposure moderation effects attained significance at alpha
poly. Failing to include all covariate-by-polymorphism and covariate-by-moderator terms as covariates, as is common in the G×E literature (
33), inflated product term test statistics on average but did not result in any additional significant effects (see section S10 of the
online supplement). Complete results for all outcomes are provided in sections S7–S10 of the
online supplement.
Despite the lack of evidence for G×E effects, all moderators exhibited large significant effects on all outcomes in the expected directions (see section S6 of the online supplement). For example, experiencing childhood trauma increased odds for estimated lifetime depression diagnosis by a factor of 1.655 (z=32.048, p=2.33×10−225) and experiencing a traumatic event in the past 2 years increased incidence rate of current depression severity index by a factor of 1.431 (z=27.004, p=1.32×10−160).
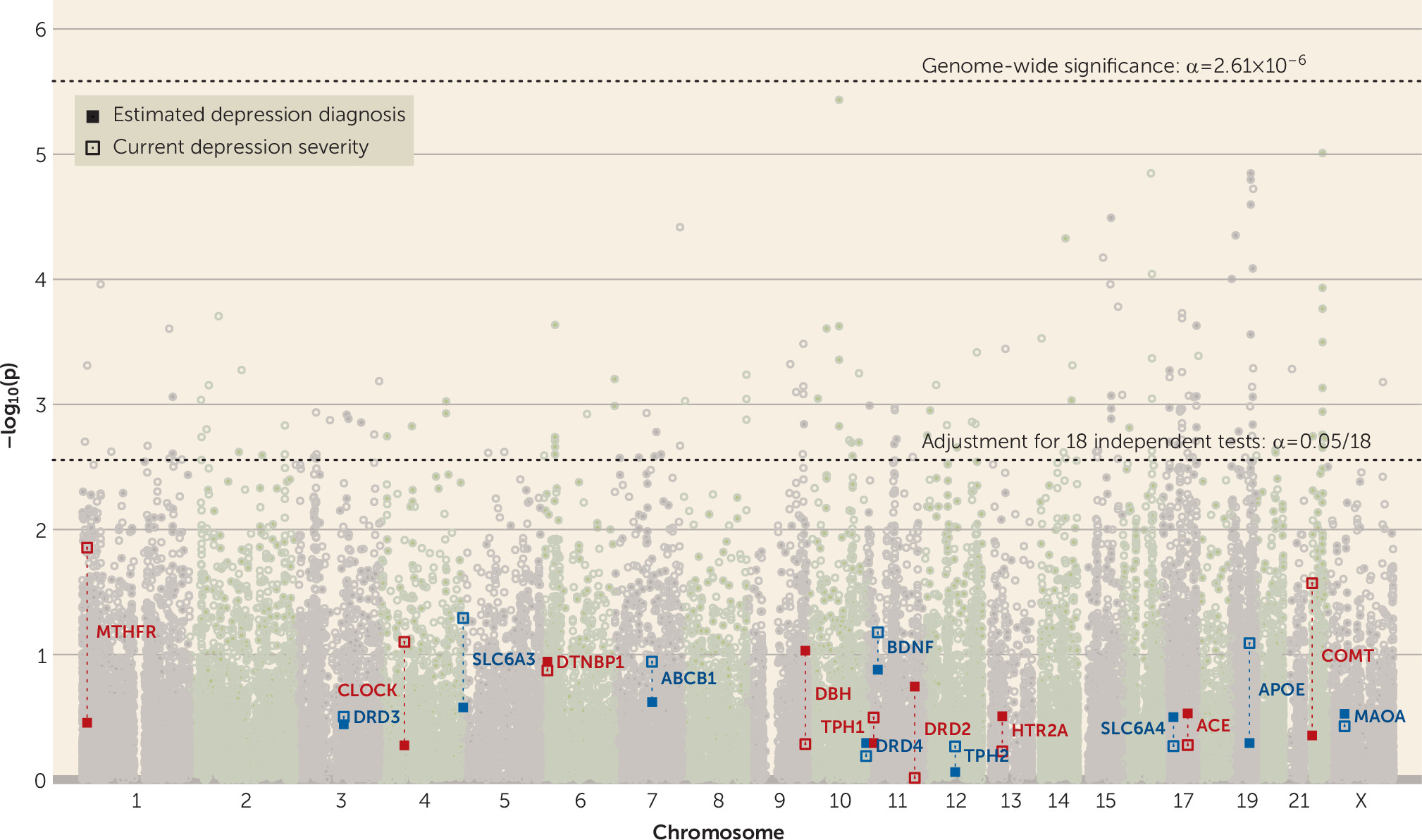
Gene-Level Analyses
Across all candidate genes and outcomes, only
DRD2 showed a significant gene-wise effect (alpha
gene=0.05/18
=2.78×10
−3), and only on PGC lifetime depression diagnosis, using both the sum −log p statistic (p
=5.14×10
−7) and the minimum p-value statistic (p
=2.74×10
−3; see
Figure 3 for gene-wise effects on estimated lifetime depression diagnosis and current depression severity, section S11.1 of the
online supplement for all gene-wise results, and section S4.2 for comparison of methods). The former estimate, based on the sum −log p statistic, was also significant at the more stringent genome-wide level (alpha
GW=0.05/19,165=2.61×10
−6).
DRD2 did not exhibit a significant effect on any of the UK Biobank outcomes despite its high genetic correlations with the UK Biobank depression phenotypes (see Table S3.3 and Figure S3.3 in the
online supplement). Investigating the effects of the 18 genes together as a set revealed no associations with depression above what would be expected by chance under the null hypothesis; the set of 18 depression candidate genes did not show stronger associations with any depression phenotype compared with all other genes at an alpha of 0.05 (see section S11.2 of the
online supplement).
Attempted Replication of Top 16 Loci Implicated by PGC GWAS Results
In order to contextualize the lack of replication of the 16 candidate genetic polymorphisms, we sought to replicate the top 16 independent genome-wide significant loci implicated for PGC lifetime diagnosis by examining their associations with estimated lifetime diagnosis in the independent UK Biobank sample (for details, see section S4.5 of the online supplement). Three loci attained significance at alphapoly (0.05/16) (rs12552, rs12658032, and rs11135349; see section S12 of the online supplement), which is consistent with the low power to detect small associations; the median power for the 16 loci was 0.143, and the 95% confidence interval for number of replications we would expect given power estimates was 2 to 7 (see Figure S4.6 in the online supplement).
Sensitivity of Results to Measurement Error
One possible reason candidate gene polymorphism associations detected in small samples are not replicated in large GWASs is the potentially worse phenotyping and higher measurement error in predictor or outcome variables in GWAS data sets. To investigate this possibility, we used a Monte Carlo procedure to quantify the extent to which measurement error may have affected the statistical power of our tests. As a lower bound on a candidate gene polymorphism study effect sizes, we used the minimally detectable log odds ratio for both main and interaction effects corresponding to 50% power at an alpha of 0.05 in a balanced case-control study of 1,000 individuals and where the risk allele frequency was 0.5 (e.g., for main effects, genomic relative risk=1.16). Simulations demonstrated that we had ∼100% power to detect such effects under multiple severe measurement error scenarios in a sample size typical of that in our UK Biobank analyses (∼30,000 case subjects and ∼85,000 control subjects; see section S4.3.3 of the online supplement). This was true even in the extreme scenario in which half of diagnoses and half of traumatic exposures were determined by coin toss (see Figure S4.5 in the online supplement).
Discussion
We examined multiple types of associations between 18 highly studied candidate genes for depression and multiple depression phenotypes. The study was very well powered compared with previous candidate gene studies, with Ns ranging from 62,138 to 443,264 across subsamples. Despite the high statistical power, none of the most highly studied polymorphisms within these genes demonstrated substantial contributions to depression liability. Furthermore, we found no evidence to support moderation of polymorphism effects by exposure to traumatic events or socioeconomic adversity. We also found little evidence to support contributions of other common polymorphisms within these genes to depression liability, except DRD2, which showed a genome-wide significant gene-wise effect on depression diagnosis in the PGC sample but not on any outcomes in the UK Biobank sample. The reasons for the failure of DRD2 to replicate in the UK Biobank are unclear, but it could be due to sampling variability, lower statistical power in the UK Biobank, or false positive or negative findings. Phenotypic heterogeneity, however, is an unlikely explanation, as genetic correlation estimates between depression phenotypes across samples were high (see Table S3.3 and Figure S3.3 in the online supplement)—for example, PGC lifetime depression diagnosis was strongly associated with estimated lifetime depression diagnosis from the UK Biobank online follow-up questionnaire (ȟ2LDSC=0.085, SE=0.004, and ȟ2LDSC=0.057, SE=0.007, respectively; řg= 0.855, SE=0.054, p=2.08×10−57), which was in turn strongly associated with probable lifetime diagnosis from the UK Biobank initial touchscreen interview (ȟ2LDSC=0.090, SE=0.008; řg=0.939, SE=0.082, p=2.83×10−30). Finally, as a set, depression candidate genes were no more related to depression phenotypes than noncandidate genes. Our results stand in stark contrast to the candidate gene literature, where large, statistically significant effects are commonly reported for the specific polymorphisms in the 18 candidate genes we investigated here.
Several features of this investigation set it apart from previous candidate gene replication attempts, meta-analyses of candidate gene studies, and genome-wide studies that failed to support roles for depression candidate polymorphisms. First, this is the only study to have imputed and examined the effects of several highly studied VNTR polymorphisms in a large GWAS data set, including 5-HTTLPR in
SLC6A4, which was examined in 38.14% of the depression candidate gene studies we identified (see reference
29 for imputation details). Second, we thoroughly examined several distinct depression phenotypes (e.g., diagnosis, depressive episode recurrence, symptom count among depressed individuals) to ensure that our results did not reflect a single operationalization of depression. Some researchers have attributed the poor replicability of candidate gene findings to specificity of effects with respect to particular types of depression or stressors (e.g., prior versus subsequent depression onset with respect to stress exposure [
38], recurrent versus single-episode depression [
55], and financial versus other stress exposure [
56]). We therefore examined all available depression and exposure phenotypes reflecting constructs of interest in the candidate gene literature. Results for all measures and modeling choices (e.g., multiplicative versus additive interactions), presented in detail in the supplement (see sections S7–S11 of the
online supplement), were consistently null with respect to candidate gene hypotheses. Third, we employed exceedingly liberal significance thresholds (e.g., for polymorphism-wise analyses, alpha
poly=3.13×10
−3, as opposed to the standard alpha
GWAS=5×10
−8 utilized in GWASs) across all outcomes to ensure that no possible effect was missed, correcting only for the number of polymorphisms we examined. Our results therefore suggest that the zero or near-zero effect sizes of these candidate polymorphisms, rather than the multiple-testing burden imposed by genome-wide scans, account for the previous failures of large GWASs to detect candidate polymorphism effects. Finally, and perhaps most importantly, unlike meta-analyses that use previously published candidate gene findings, our results cannot be affected by selective publication or reporting practices that can inflate type I errors and lead to biased representations of evidence for candidate gene hypotheses.
Our study has several limitations. First, it is possible that we failed to identify a small number of candidate gene publications and that this resulted in the omission of some depression candidate genes examined in 10 or more publications. Nevertheless, the top nine of the 18 identified genes accounted for 86.59% of the estimated number of studies, and it is unlikely that we omitted any depression candidate genes with popularity approaching that of, for example,
SLC6A4 or
COMT. Second, a subset of the UK Biobank sample was ascertained for smoking behaviors (the BiLEVE study [
57]), and controlling for genotyping batch (which differentiates the two subsamples) has the potential to induce collider bias (
58). However, only one of the 16 candidate gene polymorphisms demonstrated minor allele frequency (MAF) differences across these two subsamples (rs6311; χ
2=12.558, df=2, p=0.002; MAF=0.402 in the BiLEVE sample, MAF=0.405 otherwise) and it is unlikely that ascertainment in the BiLEVE subsample unduly influenced association statistics. However, the potential influence of ascertainment in the BiLEVE subsample on interaction effect estimates, as well as other possible sources of selection-induced bias, remains unclear. Third, whereas some of phenotypes we examined closely matched standard diagnostic instruments (e.g., current depression severity was based on the widely used Patient Health Questionnaire–9 [
59]), others were of undetermined reliability. For example, one of the nine DSM-5 depression symptoms (motor agitation/retardation) was omitted from the UK Biobank online mental health follow-up questionnaire, and our estimated lifetime depression diagnosis phenotype required four or more of eight symptoms rather than the standard five or more of nine symptoms (in addition to episode duration and impairment criteria; see section S3.1 of the
online supplement). However, enforcing stricter case-control criteria (i.e., comparing individuals who endorsed no 2-week period of either anhedonia or depressed mood throughout their lifetime to individuals reporting recurrent episodes, endorsing five or more of eight symptoms, and meeting duration and impairment criteria) failed to alter results (see sections S7–S9 of the
online supplement), despite the fact that even this diminished sample size (N=67,304) was much larger than any previous candidate gene study we are aware of. Fourth, some of the phenotypes we examined were possibly measured with greater error than is typical in smaller candidate gene studies, an issue for which large studies are often criticized. For example, the prevalence of our measure of traumatic exposure in adulthood was uncommonly high (59.11%), and most of our retrospective measurements were likely corrupted by recall bias. However, as demonstrated in section S4.3.3 of the
online supplement, even extreme measurement error cannot explain our failure to detect the relatively large effects necessary for detection in smaller samples. Furthermore, follow-up analyses demonstrated strong effects of all environmental moderators across all outcomes (see section S6 of the
online supplement), suggesting that both moderators and depression phenotypes were measured with sufficient accuracy to detect known environmental effects. It is exceedingly difficult to construct a plausible measurement error model that could, for example, comfortably reconcile the large effect estimate of childhood trauma on estimated lifetime diagnosis (odds ratio=1.655, p=2.33×10
−225) and the negligible estimate for the 5-HTTLPR-by-childhood trauma interaction effect (odds ratio=0.988, p=0.919) with the existence of a substantial G×E interaction effect.
The genetic underpinnings of common complex traits such as depression appear to be far more complicated than originally hoped (
60,
61), and large collaborative efforts have not supported the existence of common genetic variants with large effects on depression liability (
18). In the context of our understanding of psychiatric genetics in the 1990s and early 2000s, the most studied candidate genes and the polymorphisms within them were defensible targets for association studies. However, our results demonstrate that historical depression candidate gene polymorphisms do not have detectable effects on depression phenotypes. Furthermore, the candidate genes themselves (with the possible exception of
DRD2) were no more associated with depression phenotypes than genes chosen at random. The present study had >99.99% power at alpha
GWAS=5×10
−8 to detect a main effect of the magnitude commonly reported in candidate gene studies, even allowing for extreme measurement error in both outcome and moderator phenotypes (see section S4.3 of the
online supplement). Thus, it is extremely unlikely that we failed to detect any true associations between depression phenotypes and these candidate genes. The implication of our study, therefore, is that previous positive main effect or interaction effect findings for these 18 candidate genes with respect to depression were false positives. Our results mirror those of well-powered investigations of candidate gene hypotheses for other complex traits, including those of schizophrenia (
16,
25) and white matter microstructure (
19). The potential for self-correction is an essential strength of the scientific enterprise; it is with this mechanism in mind that we present these findings. In agreement with the recent recommendations of the National Institute of Mental Health Council Workgroup on Genomics (
62), we conclude that it is time for depression research to abandon historical candidate gene and candidate gene-by-environment interaction hypotheses.
Acknowledgments
The authors thank SURFsara (
www.surfsara.nl) for support in using the Lisa Compute Cluster. They also thank the research participants of the PGC and UK Biobank, and the employees of 23andMe for their contribution to this study.