Progesterone is highly lipophilic and easily passes through the blood-brain barrier (
9). Animal studies and postmortem studies in reproductive and postmenopausal women indicate that progesterone is accumulated in the brain (
10–
12), with the highest concentrations found in the amygdala (
10). Preclinical data suggest that progesterone receptors are found throughout the amygdala, hippocampus, hypothalamus, thalamus, and frontal cortex (
13–
17), regions of importance in emotion processing. Although the exact mechanism by which progesterone precipitates the symptoms of PMDD is unknown, interactions with the serotonin (
18,
19) and the GABAergic systems (
20,
21) are plausible (
22).
Ulipristal acetate (UPA) is a selective progesterone receptor modulator that acts as a progesterone antagonist in progesterone-responsive tissues, including the brain (
23). UPA binds to progesterone receptors A and B with high affinity (
24,
25) and interferes with progesterone receptor–mediated DNA transcription (
26). UPA, in the dose and regimen employed in this study, is used for treatment of uterine fibroids (
27). Low-dose continuous UPA treatment leads to anovulation in approximately 80% of women with uterine fibroids (
23); this effect and progesterone receptor antagonism are the suspected mechanisms for symptom relief in women with PMDD. Thus, the aim of this proof-of-concept study was to investigate the usefulness of UPA for treatment of PMDD. We hypothesized that overall premenstrual PMDD symptoms would improve in the group treated with UPA compared with the placebo group, with efficacy being more marked for the mental symptoms of irritability and depression.
Methods
Participants
The study was carried out at the Departments of Obstetrics and Gynecology at Uppsala University Hospital, Karolinska University Hospital, Danderyd University Hospital, and Umeå University Hospital between January 15, 2017, and October 19, 2019. Because of the European Medical Agency’s Pharmacovigilance Risk Assessment Committee review of the study drug, recruitment was temporarily stopped between February 9, 2018, and August 13, 2018. We recruited participants by advertisements in local newspapers and social media.
Women were eligible if they were 18–46 years of age, healthy, had regular menstrual cycles, and met criteria for PMDD. We excluded women who had ongoing mental health problems as assessed with the Mini International Neuropsychiatric Interview (MINI) (
28), ongoing drug or alcohol abuse, past hospitalization for a psychiatric disorder or attempted suicide, or treatment with psychotropic medication during the previous 3 months. In addition, we excluded women with severe medical conditions, including liver disease, women who had been treated with hormonal contraceptives or any other steroid hormone treatment during the previous 3 months, women who were breastfeeding, pregnant, or planned a pregnancy, and women with abnormal liver function tests.
Women with past depressive and anxiety disorders were allowed to participate in the study. Past depressive episodes and past panic disorder were captured by the MINI. Past anxiety disorders and treatment were assessed with structured questions in the case report form.
We used the Daily Record of Severity of Problems (DRSP), filled out daily for at least two menstrual cycles using a smartphone application, for the PMDD diagnosis (
29). The DRSP consists of 21 items that reflect the 11 candidate symptoms for PMDD, depression, anxiety, mood swings, irritability, decreased interest in usual activities, difficulties concentrating, fatigue, sleep disturbances, increased appetite or cravings, sense of being overwhelmed, and physical symptoms. Each item is scored on a 6-point Likert scale ranging from 1 (not at all) to 6 (extreme).
According to DSM-5, PMDD is defined as an increase >50% in at least five of 11 symptoms between the follicular (days 6–12) and luteal phase (days −7 to −1). Among the five symptoms, at least one must be a core symptom (
30). Percent increase was calculated as (mean luteal phase scores − mean follicular phase scores / mean follicular phase scores) × 100. We required that diagnostic symptoms be at least mild (mean luteal phase score >3.0) and disappeared during the follicular phase (mean follicular phase score <2.0). If women did not meet criteria for PMDD after 2 months of charting, they were allowed to score symptoms for an additional cycle. However, only two women who needed a follow-up cycle were eventually included. The majority of women who needed a follow-up cycle did not meet diagnostic criteria. Additionally, as part of the daily diary, women were asked to report on menstrual bleeding.
The women received oral and written information and had the opportunity to ask questions before signing informed consent in the presence of an investigator. They were not reimbursed. The study procedures were conducted in accordance with Good Clinical Practice and the Helsinki Declaration’s ethical standards for human experimentation. The study was approved by the Regional Ethical Review Board (2016/184), Uppsala, and the Medical Products Agency in Sweden. The clinical trial identifier is EudraCT 2016-001719-19.
Study Design and Procedures
The study was an investigator-initiated, multicenter, double-blind, randomized, parallel-group clinical trial in which participants were treated with either 5 mg/day of UPA or placebo during three 28-day treatment cycles. Women were randomly assigned to these treatments in a 1:1 ratio.
Screening, Study Visits, And Assessments.
Eligible women first attended a screening visit, during which demographic data and information on previous, recent, and ongoing medication were collected. After two diagnostic cycles, eligible women made a second visit (baseline/randomization), scheduled in the late luteal phase. At this visit, they filled out the EuroQoL visual analogue scale (EQ-VAS) (
31) and the Montgomery-Åsberg Depression Rating Scale, self-rated version (MADRS-S) (
32). After randomization, women started taking the study drug on the first day of menses. Participants were not informed that UPA could alter their menstrual cycles. The last and third visit was made during the premenstrual phase of the third treatment cycle or, for women who developed irregular menses or lost their menses, during the final week of active treatment. At the last visit, participants filled out the EQ-VAS and MADRS-S once again. A blood sample was collected at baseline and during the final visit for hormone analyses. In between the face-to-face meetings, the study nurses were in contact with the patients by telephone during the first treatment cycle. Any adverse events or change in concomitant medication were queried and registered at each visit. The study nurses followed the daily symptom charts and contacted patients who failed to chart symptoms for more than 3 consecutive days. As of February 9, 2018, all women made an additional visit during each treatment cycle for liver function tests.
UPA and Placebo.
UPA and identical-looking placebo tablets were provided by Gedeon Richter Nordics AB. Apoteksbolaget Production and Laboratories (the National Corporation of Swedish Pharmacies) prepared the randomization lists, using a computerized random number generator in blocks of four, which was blinded to the local investigators. Recipharm AB (Stockholm) did the packaging and labeling of study drugs. At randomization, participants received the numbered container with the lowest available number. During the study, participants and study personnel were not informed about which treatment the patient received, and randomization codes were kept at Recipharm AB until the study was completed. Upon completion of the study, participants brought back empty containers and any remaining tablets were counted by the study nurses.
Hormone Analyses.
Venous blood samples were collected from each participant to determine the levels of estradiol and progesterone. Steroid hormones were measured in serum at the Core Facility of Metabolomics at the University of Bergen by liquid chromatography–tandem mass spectrometry. Sample processing was robotized (Hamilton STAR) and included protein precipitation with acetonitrile and liquid-liquid extraction with ethyl acetate-heptane. The samples were analyzed on an Acquity UPLC system (Waters, Milford, Mass.) connected to a Xevo TQ-S tandem mass spectrometer (Waters). The compounds were separated on a C-18 column (50×2.1 mm, 1.7-mm particle size), which is developed by gradient elution over 14 minutes, using water and methanol containing ammonium hydroxide as mobile phases. They were detected in negative (e.g., estradiol) or positive ion (progesterone) MRM mode. Two product ions are monitored for each compound to check for interferences. The method is validated for estradiol (sensitivity, 3.6 pmol/L [lower limit of quantitation] and 1.2 pmol/L [level of detection] and total control volume [coefficient of variation] for intermediate concentrations, 5.0%) and progesterone (10.3%). Because of the COVID-19 pandemic, only 70 samples at baseline (36 from the UPA treatment group) and 46 samples at follow-up (22 from the UPA group) could be analyzed.
Outcomes
We anticipated that women assigned to UPA treatment could develop amenorrhea or sparse menstrual cycles (>35 days between menses). Thus, treatment outcomes are reported in relation to premenstrual periods rather than treatment cycles. We evaluated treatment outcomes during 5 premenstrual days of the two last menstrual cycles of each participant during the study period (here referred to as the penultimate and last premenstrual periods). In women who had no menses during the study period, the final 5 days of treatment cycles 2 and 3 were used—that is, the approximate days of their luteal phases had their menses continued to be regular.
The primary outcome measure was the change from baseline in premenstrual DRSP total score. We generated the DRSP total score by computing the mean of each item during the final 5 days of the premenstrual phase of the baseline and treatment cycles, and summed the 21 items.
Secondary outcomes were change from baseline in three DRSP subscales, in the individual DRSP symptoms, and the three DRSP functional items. The DRSP depressive symptom subscale consists of the symptoms felt depressed, felt hopeless, felt worthless or guilty, slept more, had trouble sleeping, and felt overwhelmed; the anger/irritability subscale consists of the symptoms anger and/or irritability and conflicts with others; and the physical symptom subscale consists of the symptoms breast tenderness, bloating, headache, and joint or muscle pain. The subscales and individual symptoms, including the functional items, from the DRSP were scored on the same days as the DRSP total score.
We also evaluated PMDD remission at the final treatment cycle, based on the mean premenstrual DRSP scores from this cycle. In line with the diagnostic criteria for PMDD, complete remission was defined as no symptom with a mean premenstrual score >3.0 during the final treatment cycle. Partial remission was defined as having one to four symptoms with a mean luteal phase score >3.0 during the final treatment cycle. Because of many prolonged menstrual cycles, we refrained from using change from follicular phase in the remission criteria.
Secondary outcomes also included self-rated depressive symptoms (MADRS-S) and quality of life (EQ-VAS). The MADRS-S inquires about nine symptoms of depression, each rated on a six-point scale (e.g., with regard to inability to feel: 0=normal interest in surroundings/other people; 2=reduced ability to enjoy usual interests; 4=loss of interest in surroundings/loss of feelings for friends; 6=emotionally paralyzed, unable to feel anger, grief, or pleasure; complete failure to feel for close relatives and friends), yielding a maximum score of 54. Compared with other depression rating scales, the psychometric profile of the MADRS-S is excellent for detecting change in depressive symptoms (
33). We compared the change from baseline in luteal phase MADRS-S scores between treatment groups.
The EQ-5D is the most widely used generic patient-reported outcome questionnaire internationally, capturing information on mobility, self-care, usual activities, pain/discomfort, and anxiety/depression (
34). For this relatively young population, we used the EQ-VAS, which measures current health status on a scale from 0 to 100, where 0 is the worst imaginable health state and 100 is the best. We compared the change from baseline in luteal phase EQ-VAS ratings between treatments.
Statistical Analysis
The power analysis was based on other strategies that are used to inhibit ovulation in women with PMDD (
35). Based on a difference of 1.5 of the core DRSP symptoms and a standard deviation ranging between 1.8 and 2.0, we had more than 90% power to detect a difference between treatments after recruiting 75 subjects. As for treatment response, the study had sufficient power (>80%) to detect a difference between treatments, assuming complete or partial response in 75% of women randomized to UPA and 40% remission in the placebo group. As we assumed a 25% dropout rate, we planned to recruit 100 women.
Primary Outcomes.
We used an intention-to-treat concept—that is, women were retained in the analyses if they had sufficient data—and primary analyses were performed with unblinded data. We used linear mixed-model analysis of variance to evaluate the treatment effect on DRSP total score. Participants were entered as subjects, with time point (baseline and the last two premenstrual periods) as a repeated variable, and using the first-order autoregressive repeated covariance type. Treatment (UPA or placebo) was entered as a fixed factor, and the maximum likelihood estimation method was used. The interaction term between treatment and time was included to assess the effect of treatment. We calculated mean difference and 95% confidence intervals for the DRSP total score for the last two premenstrual periods.
Secondary Outcomes.
We used the same approach for the DRSP summed subscales and individual symptoms, the MADRS-S scores, and the ED-VAS scores as we did for the DRSP total score.
Chi-square tests were used to compare the treatment response between UPA and placebo. Correction for multiple testing was applied to the DRSP subscales (three tests), the DRSP single items (21 tests), and the functional subscales (three tests).
SPSS (IBM, Armonk, N.Y.) was used for the analyses, and p values <0.05 were considered statistically significant.
Results
Study Population
In total, 95 women with PMDD underwent randomized assignment to either UPA (N=48) or placebo (N=47) (see Figure S1 in the online supplement). Seventeen women discontinued (eight in the UPA group and nine in the placebo group; the dropout rate was 17.9%), most often during the first treatment cycle (eight in the UPA group and five in the placebo group). Seven women in the UPA group discontinued because of mild or moderate side effects. The most commonly reported side effects that led to discontinuation in the UPA group were headache, fatigue, and nausea, but one woman discontinued because of worsening of depressive symptoms. Among women in the placebo group, three discontinued because of depressive symptoms or anxiety. Two of the women who dropped out had data that could be included in the primary outcome analyses. According to hormonal levels measured at baseline, women were assessed during the late luteal phase (mean estradiol levels, 416 pmol/L [SD=287], and mean progesterone level, 17.8 nmol/L [SD=15.0]), with no significant differences between groups. As expected, the women in the UPA group had lower progesterone levels (5.8 nmol/L, SD=9.9) than those in the placebo group (15.0 nmol/L, SD=16.3) at the last premenstrual period (t=2.3, df=44, p=0.03), while estradiol levels remained in the midfollicular range (488 pmol/L [SD=418] compared with 402 pmol/L [SD=267]; t=−0.83, df=44, p=0.41).
Among women who completed the study, 6 days (SD=11) were missing in the daily diaries, mostly outside the premenstrual phase. Two women with more than 40% missing days were excluded from the analyses that incorporated the DRSP ratings. Thus, 41 women in the UPA group and 39 the placebo group were included in the analyses. Adherence to treatment was good, with 0–5 remaining tablets at the final visit among women who completed the trial. Among women in the UPA group who completed the study, 11 (27.5%) had no menses during the study period, 23 (57.5%) had sparse menses (i.e., more than 35 days between menses), and six (15.0%) maintained regular menses.
The demographic and clinical characteristics of the study population are summarized in
Table 1. The mean age of women in the placebo and UPA groups was 35.0 years (SD=6.4) and 36.6 years (SD=5.7), respectively. The majority of women were married or cohabiting, were working part-time or full-time, and had a high education level. The majority had a history of depressive episodes or anxiety disorders, and more than two-thirds had previously been treated for PMDD. Among women who had previously used antidepressants, 35.7% had stopped because of nonresponse. We found no significant differences between the two treatment groups in demographic and clinical variables.
Primary Outcome
Primary and secondary treatment outcomes are presented in
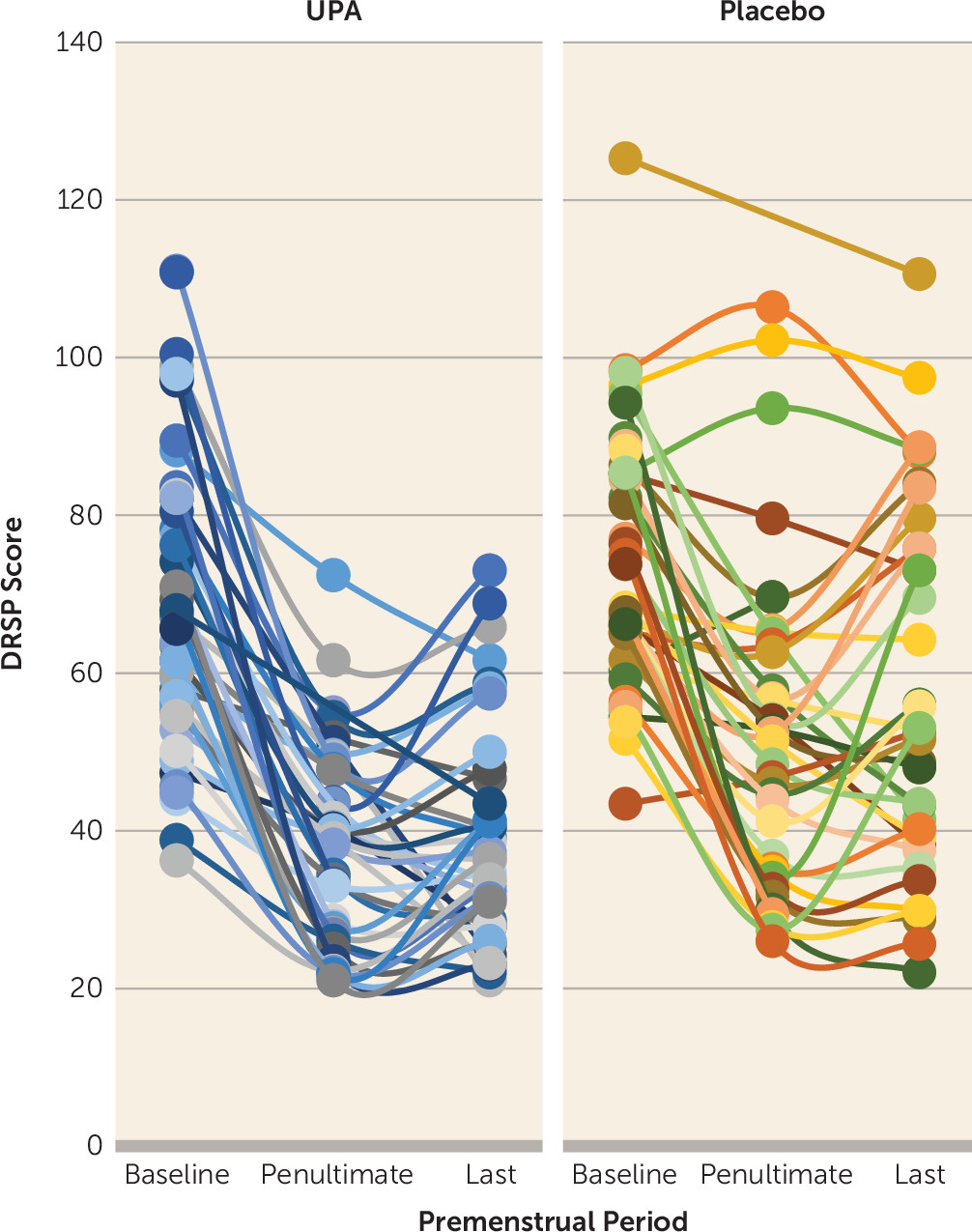
Table 2. We found significant treatment-by-time interactions in favor for UPA treatment for DRSP total score (F=6.02, df=2, 134.8, p=0.003). The mean improvement in DRSP total score at the penultimate premenstrual period was 43% (SD=17) in the UPA group, compared with 27% (SD=23) in the placebo group (mean difference, −16%, 95% CI=−25, −6). The corresponding numbers during the last premenstrual period were 41% (SD=18) and 22% (SD=27) in the UPA and placebo groups, respectively (mean difference, −18%, 95% CI=−29, −8).
Figure 1 illustrates DRSP scores across the premenstrual periods of the study.
Secondary Outcomes
We noted significant treatment-by-time interactions for the DRSP depressive symptom subscale (F=4.92, df=2, 133.0, p=0.009) and the DRSP anger/irritability subscale (F=8.53, df=2, 140.3, p<0.001), but not for the DRSP physical symptom subscale (F=0.72, df=2, 142.6, p=0.5) (
Table 2). The treatment effects were observed by the penultimate premenstrual period on the DRSP depressive symptom subscale (mean difference between treatments in change from baseline, −2.8, 95% CI=−5.2, −0.3) and on the DRSP anger/irritability subscale (mean difference between treatments in change from baseline, −1.7, 95% CI=−2.8, −0.7) (
Table 2). In the last premenstrual period, the mean difference in change from baseline was −3.4 (95% CI=−6.1, −0.8) for the DRSP depressive symptom subscale and −1.8 (95% CI=−2.9, −0.7) for the DRSP anger/irritability subscale (
Table 2).
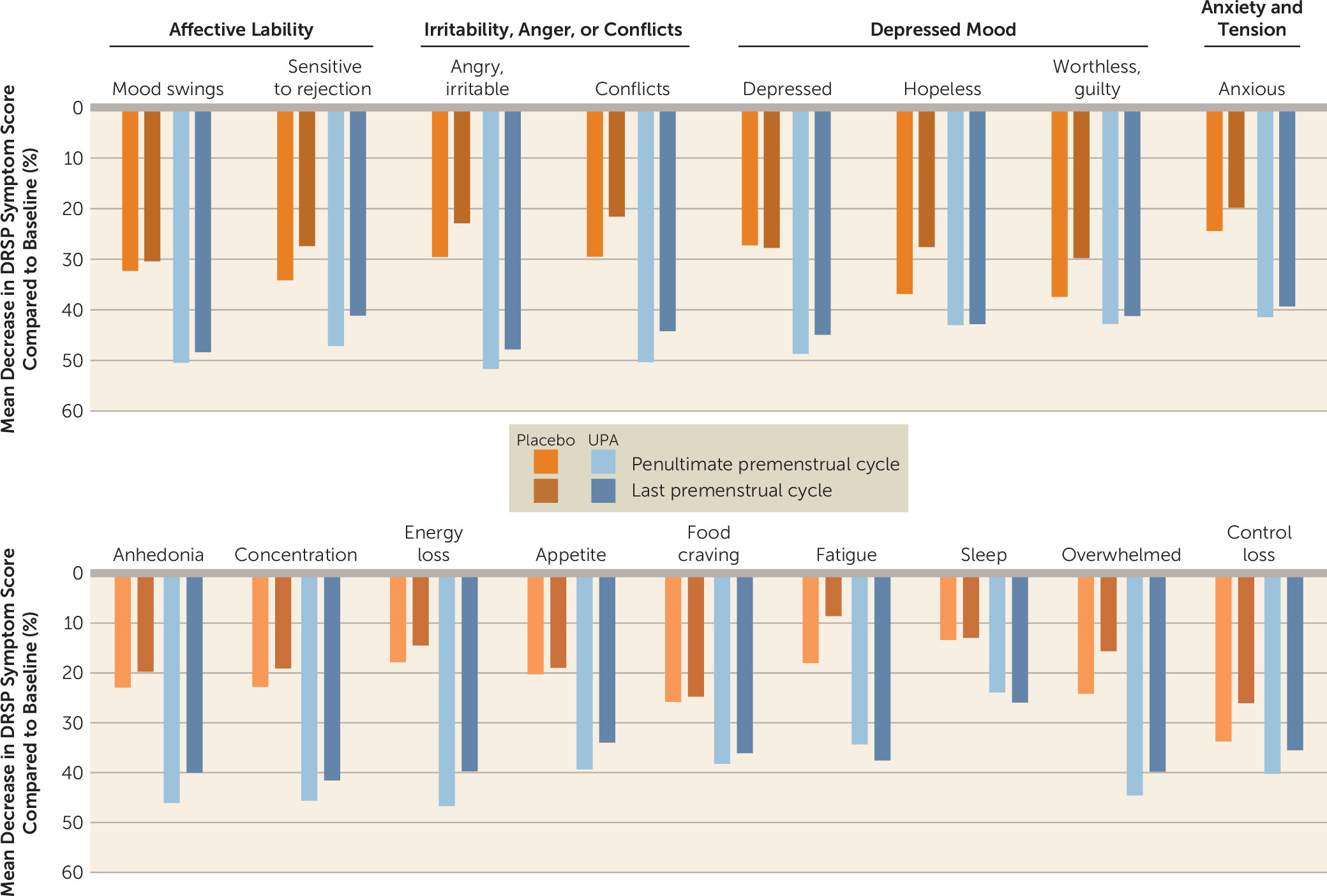
We found significant treatment effects, favoring UPA, for a number of individual symptoms on the DRSP scale, although some items did not remain significant after correction for multiple testing: felt depressed (F=6.98, df=2, 140.0, p=0.001), angry/irritable (F=8.38, df=2, 135.5, p<0.001), had conflicts or problems with others (F=6.92, df=2, 144.8, p=0.001), and lack of energy (F=9.28, df=2, 126.7, p<0.001) (
Table 2). For most of these symptoms, treatment effects were noticeable by the penultimate premenstrual period (
Figure 2). The UPA and placebo groups did not differ in physical symptom severity from baseline to end of study (all p values >0.05) (
Table 2).
We also noted treatment effects, favoring UPA, in the DRSP functional items, namely, social function (F=4.96, df=2, 129.0, p=0.008) and family function (F=4.60, df=2, 140.9, p=0.012) (
Table 2).
Complete or partial remission during the last premenstrual period was attained by 20 women (50.0%) and 14 women (35.0%), respectively, in the UPA group, whereas the corresponding numbers in the placebo group were eight women (21.1%) and 12 women (31.6%), respectively (χ2=11.2, p=0.004).
Women in both treatment groups had lower MADRS-S and higher EQ-VAS scores at the final visit, but no effect of UPA treatment was demonstrated (MADRS-S score, F=0.14, df=1, 74, p=0.7; EQ-VAS score, F=2.09, df=1, 73, p=0.2) (
Table 2).
Side Effects
Side effects were rare (see Table S1 in the online supplement). The most commonly reported side effects among women in the UPA group were headache (8.3%), nausea (8.3%), and fatigue (6.3%). Nausea was significantly more common among women in the UPA group than in the placebo group (χ2=4.1, p=0.043). No other differences in side effects were noted. No serious adverse events occurred, and none of the women had abnormal liver function tests at any point during the study.
Discussion
The present findings suggest that UPA is an effective treatment for PMDD, and particularly for the mental symptoms associated with the syndrome. We found significant treatment effects for the DRSP total score and the depressive symptom and anger/irritability subscales. Complete or partial remission was attained by 85% of women in the UPA treatment group. Improvements were noted in quality of life and self-reported depressive symptoms in both groups, although the improvement did not differ significantly between the treatment groups.
No previous studies have investigated UPA for use in PMDD, but the results are not surprising given that continuous UPA treatment leads to anovulation in the majority of women (
23). Previous attempts to use treatments that induce anovulation, such as GnRH agonists, have been successful in alleviating both mental and physical PMDD symptoms (
3,
4). However, the usefulness of GnRH agonists is limited by their hypoestrogenic side effects, which in the short term are experienced by the women as vasomotor symptoms, and in the long term may lead to bone demineralization (
3,
4). In contrast, with UPA treatment, estradiol serum concentrations are maintained at mid-follicular-phase levels (
23), and women on UPA rarely suffer from vasomotor symptoms (
36). This study also confirms the beneficial side effect profile of the compound and the maintained estradiol levels (
26,
27,
36).
Two small randomized clinical trials conducted in the 1990s with a progesterone receptor antagonist were likely compromised by the administration of the drug days after ovulation, which is too late in the menstrual cycle to have any effect (
37,
38).
While UPA was effective in treating the mental symptoms of PMDD, we found no beneficial effects on the physical symptoms. This is in contrast with the GnRH agonists and certain combined hormonal contraceptives, which are helpful also for breast tenderness, bloating, and other physical symptoms associated with the syndrome (
3,
39). Reasons for the absence of treatment effect on physical symptoms may reside in the study population, mainly characterized by high scores on mental symptoms, or progesterone receptor agonist effects in certain tissues, or insufficient suppression of estradiol levels (
23). Moreover, in contrast to the emotional symptoms, somatic symptoms have also shown limited improvement in studies of serotonin reuptake inhibitors with various dosing regimens (
40,
41). Physical discomfort may, however, be better tolerated in the presence of diminished irritability and dysphoria.
The strengths of the study include its being a multicenter randomized controlled trial and the use of daily diaries for capturing both menses and mental symptoms throughout the study. Adherence was excellent, both with treatment and with daily symptom chartings. Another strength is the three-cycle duration, which demonstrates the persistence of response and the viability of the treatment over time. The use of one primary outcome measure (i.e., change from baseline in premenstrual DRSP total score) in an adequately powered sample is another strength. Given that this was a proof-of-concept study, the findings related to the secondary outcomes were presented according to an exploratory approach, although some were not significant after correction for multiple testing.
Limitations of the study include the likely possibility that blinding was compromised, as UPA treatment in some patients led to irregular menses or amenorrhea. While intrinsic to the pharmacological effects of UPA, this problem is shared with studies of GnRH agonists and some hormonal contraceptives. Even though women were not informed that UPA could lead to amenorrhea, they may have read about UPA and its side effects. Future studies should include a larger sample to investigate symptomatic benefit separately among the women who remain ovulatory.
As this study was not designed to capture ovulation and progesterone levels during treatment, we cannot ascertain whether the effect of UPA is mediated by anovulation, leading to low progesterone and allopregnanolone levels, or via specific actions at the progesterone receptor. However, we note that the proportion of women who developed amenorrhea in response to UPA was much lower (27.5%) than in studies on uterine fibroids, where amenorrhea occurs in approximately 80% of women (
26,
31,
42). For this reason, we speculate that both factors are likely involved—that is, lowered progesterone levels and specific actions at the receptor in women who maintained menstrual cycles.
Moreover, as estradiol levels are maintained at mid-follicular-phase levels, the effect of UPA may also comprise estrogen’s effects on mood. In line with this, menopause-like pharmacologically induced estrogen levels have been associated with depressive symptoms (
43). While not discerning the effect of these hormones separately, after GnRH agonist treatment, PMDD symptom reinstatement has indeed been demonstrated with estradiol and progesterone add-back (
4,
8). Thus, it is plausible that UPA contributes to PMDD symptom relief by maintaining low and constant levels of both progesterone and estradiol. Clearly, further studies are needed to elucidate the specific actions of UPA in women with PMDD.
Moreover, with one-third of the PMDD patients previously unsuccessfully treated with an antidepressant, symptom improvement following UPA treatment highlights this treatment as a valid alternative for patients who do not respond to selective serotonin reuptake inhibitors, but also calls for further studies to generalize the present findings to treatment-naive patients.
Regarding the study population, the mild to moderate baseline DRSP scores are in the range of previous studies (
40,
44). MADRS-S scores define the present sample as moderately depressed at baseline (scores were slightly to much lower than in samples with major depression or treatment-resistant major depression [
45,
46]). At follow-up, the scores of the UPA group describe women as having no depression but those in the placebo group as having mild depressive symptoms (
32). Similarly, in the UPA group, EQ-VAS scores meaningfully improved from indicating poor health-related quality of life to values defined as normal in population-based samples, while in the placebo group the improved health status remained well below these levels (
47). Whereas MADRS-S or EQ-VAS scores improved over time, the lack of between-group differences may be explained by the fact that they are one-time-point measurements and thus are sensitive to the daily mood of the participant. Additionally, the constellation of mood-related impairment addressed by the MADRS-S may apply only to a subset of women with PMDD and possibly not be the main target of UPA treatment, as irritability showed the highest improvement. Additionally, a greater general health-related quality of life improvement due to UPA treatment may be seen over a longer period as the patient becomes familiar with its effects and adjusts her daily life.
UPA was introduced in 2012, and more than 750,000 women have been treated for uterine fibroids with the drug (
48). Most commonly, women received a 3-month treatment course, but studies with safety and efficacy data with up to four 3-month courses have shown that side effects remain stable or even decrease with an increasing number of treatment courses (
26). During the spring of 2018, the Pharmacovigilance Risk Assessment Committee (PRAC) of the European Medicines Agency (EMA) investigated the potential role of UPA in liver injury and concluded that the drug may have contributed to some of the cases of severe liver injury that had been reported (
26). As a result, the PRAC proposed new prescription rules in order to minimize the risk of livery injury, including repeated liver function tests during the first 3 months of treatment (
49). At the same time, the U.S. Food and Drug Administration has requested additional information to be able to approve UPA in its current form. In the spring of 2020, PRAC started a new review of the drug after a single case of liver injury (
50). The results of the new review are still pending. Studies to determine the effects of UPA on the liver are ongoing, and some preliminary results, including ours, are reassuring (
51). As this proof-of-concept study is the first to evaluate UPA for treatment of PMDD, we cannot make any treatment recommendations at present. Further studies to validate our findings, and positive outcomes from ongoing liver safety studies, are needed. Last but not least, other selective progesterone receptor modulators with theoretically less effects on the liver are on their way into the market, potentially representing safer alternatives (
52).
In conclusion, UPA is a promising drug for treatment of PMDD, particularly for the psychological symptoms associated with the syndrome and as an alternative pharmacologic treatment to antidepressants for patients who do not respond or cannot tolerate selective serotonin reuptake inhibitors, the current standard of care. Moreover, the unique mechanism of action of this study, namely, modulation of progesterone receptors, provides insights into the potential molecular mechanisms underlying PMDD and its treatment. Further validating studies, as well as more reassuring information regarding the effect on liver function, are needed before this potentially highly effective treatment is made available to affected women.