Lower Availability of Mitochondrial Complex I in Anterior Cingulate Cortex in Autism: A Positron Emission Tomography Study
Publication: American Journal of Psychiatry
Abstract
Objective:
Mitochondrial dysfunction has been implicated in the pathophysiology of autism spectrum disorder (ASD) in previous studies of postmortem brain or peripheral samples. The authors investigated whether and where mitochondrial dysfunction occurs in the living brains of individuals with ASD and to identify the clinical correlates of detected mitochondrial dysfunction.
Methods:
This case-control study used positron emission tomography (PET) with 2-tert-butyl-4-chloro-5-{6-[2-(2-[18F]fluoroethoxy)-ethoxy]-pyridin-3-ylmethoxy}-2H-pyridazin-3-one ([18F]BCPP-EF), a radioligand that binds to the mitochondrial electron transport chain complex I, to examine the topographical distribution of mitochondrial dysfunction in living brains of individuals with ASD. Twenty-three adult males with high-functioning ASD, with no psychiatric comorbidities and free of psychotropic medication, and 24 typically developed males with no psychiatric diagnoses, matched with the ASD group on age, parental socioeconomic background, and IQ, underwent [18F]BCPP-EF PET measurements. Individuals with mitochondrial disease were excluded by clinical evaluation and blood tests for abnormalities in lactate and pyruvate levels.
Results:
Among the brain regions in which mitochondrial dysfunction has been reported in postmortem studies of autistic brains, participants with ASD had significantly decreased [18F]BCPP-EF availability specifically in the anterior cingulate cortex compared with typically developed participants. The regional specificity was revealed by a significant interaction between diagnosis and brain regions. Moreover, the lower [18F]BCPP-EF availability in the anterior cingulate cortex was significantly correlated with the more severe ASD core symptom of social communication deficits.
Conclusions:
This study provides direct evidence to link in vivo brain mitochondrial dysfunction with ASD pathophysiology and its communicational deficits. The findings support the possibility that mitochondrial electron transport chain complex I is a novel therapeutic target for ASD core symptoms.
Autism spectrum disorder (ASD) is a highly prevalent neurodevelopmental disorder that affects 1 in 44 children (1). The pathogenesis of ASD is not yet fully understood, and no effective medications have been approved for its core symptoms of difficulties in social interaction and communication and restricted interests and repetitive behaviors (2). Given that currently available medications are approved for the treatment only of comorbid symptoms, such as irritability, anxiety, and depression, there is an urgent need to identify molecular targets in the brain for novel pharmacological treatments of ASD itself.
Several lines of evidence from recent studies with blood or urine or with magnetic resonance spectroscopy support the idea that mitochondrial dysfunction is implicated in the pathophysiology of ASD (3, 4). Moreover, while the mitochondrial electron transport chain complex I (MC-I) genes are encoded in both the nuclear and mitochondrial genomes, postmortem studies of autistic brains have found lower nuclear gene expression of MC-I (5, 6). MC-I is the first rate-limiting step of the electron transport chain, which operates to produce energy in the form of ATP; this is responsible for neurotransmitter synthesis and synaptic plasticity (7). Because the brain has a high ATP demand, the finding of decreased MC-I indicates a role of mitochondrial dysfunction in the brain pathophysiology of ASD. More specifically, postmortem studies have found altered MC-I in the anterior cingulate cortex (ACC), superior temporal gyrus, occipital cortex, dorsolateral prefrontal cortex, thalamus, and primary motor cortex of individuals with ASD (5, 6, 8). However, no previous studies have examined mitochondrial dysfunction in the living brains of individuals with ASD, or its relationship with the core symptoms of ASD.
In vivo estimates of MC-I availability in the living human brain are now possible using positron emission tomography (PET) measurements with 2-tert-butyl-4-chloro-5-{6-[2-(2-[18F]fluoroethoxy)-ethoxy]-pyridin-3-ylmethoxy}-2H-pyridazin-3-one ([18F]BCPP-EF), a radioligand that binds to MC-I and permits investigation of the topographical distribution of mitochondrial dysfunction (9). Our previous studies confirmed that the uptake of [18F]BCPP-EF reflected the specific binding to cellular MC-I (for details, see the online supplement ). By utilizing [18F]BCPP-EF PET, the present study aimed 1) to investigate whether and where mitochondrial dysfunction occurs in the living brains of individuals with ASD, and 2) to identify the clinical correlates of detected mitochondrial dysfunction.
Methods
The protocol of this case-control study, including the main hypothesis, outcome measures, choice of imaging pre-processing, analysis, and statistical investigation, was reviewed and approved by the Ethics Committee of the Hamamatsu University School of Medicine (17-086), and was preregistered as UMIN000029183 in the University Hospital Medical Information Network. All participants provided written informed consent before participating in the study; for participants under age 20, consent from their parent was also obtained.
Participants
The inclusion criteria were age >18 years, male, ASD diagnosis based on DSM-5 criteria (2), IQ >70 as assessed with the Wechsler Adult Intelligence Scale, 3rd ed., and no neurological disorders. Individuals with ASD were recruited from among outpatients of the Hamamatsu University School of Medicine Hospital, with high priority for individuals not taking any psychotropic medication and without any psychiatric comorbidities, to minimize potential confounding effects. The diagnosis of ASD was confirmed using the Autism Diagnostic Interview–Revised (ADI-R) and the Autism Diagnostic Observation Schedule, 2nd ed. (ADOS-2). A comparison group of typically developed participants was recruited through public advertisement. To control potential confounding effects of sex differences in brain function and their interactions with ASD diagnosis, study participants were confined to males. All participants were interviewed by a trained psychiatrist (Ya.K.) to screen for the presence of psychiatric disorders using the Structured Clinical Interview for DSM-5–Research Version. Typically developed participants were excluded if they had a past or current history of psychiatric disorders. Each participant also underwent laboratory screening of peripheral blood to measure plasma lactate and pyruvate levels, to rule out mitochondrial diseases. All participants were confirmed to have no use of psychotropic medication within the previous 4 weeks and were evaluated with the 50-item Autism Spectrum Quotient, the Center for Epidemiologic Studies Depression Scale (CES-D), and the State-Trait Anxiety Inventory (STAI). Socioeconomic status was assessed in all participants with the Hollingshead index; the socioeconomic status of the participants’ parents was also assessed.
PET Data Acquisition
All participants underwent a PET scan with [18F]BCPP-EF to measure MC-I availability. After back-projection and filtering (Hanning filter, cutoff frequency of 0.2 cycles per pixel), the image resolution was 2.9×2.9×3.4 mm (full width at half maximum). The voxel of each reconstructed image measured 1.3×1.3×3.4 mm. During the scan, head movements were restricted using a thermoplastic face mask. The [18F]BCPP-EF (2 MBq/kg) was administered via an intravenous cannula as a smooth bolus injection over 10 seconds. We created parametric images of the standard uptake value (SUV) using PET data from 70 to 90 minutes after administration using a high-resolution brain PET scanner (SHR12000, Hamamatsu Photonics K.K., Hamamatsu, Japan) (10). To evaluate MC-I, we then created parametric images of the SUV ratio (SUVR), which is the ratio of regional MC-I availability to that of the whole brain. The detailed procedure for calculating SUVR has been described elsewhere (10). Briefly, the SUV of each brain region was divided by the SUV of the whole brain as the global mean of the same participant; this was then expressed as the SUVR image. Since our previous study showed a strong correlation between the quantitatively estimated value of [18F]BCPP-EF volume of the distribution and SUVR (r2=0.830, p<0.001) (10), the use of [18F]BCPP-EF SUVR was adequate for semiquantitative evaluation. These procedures were performed using PMOD, version 3.8 (PMOD Technologies, Zurich).
MRI Data Acquisition
We used a 3.0-T MRI scanner (Ingenia; Philips Healthcare, Eindhoven, the Netherlands) with three-dimensional mode sampling using the following acquisition parameters: repetition time=6.8 ms; echo time=3.1 ms; flip angle=9°; slice thickness=1.2 mm; voxel size=1.0×1.0×1.2 mm3; matrix=256×256; number of slices, 170; slice direction, sagittal. The high-resolution T1-weighted data were used to investigate possible brain structural abnormalities and to determine the regions of interest (ROIs). A trained physician (Ya.K.) located the ROIs in the target regions in subject MRI space (11). Since previous studies have shown differences in brain structure between individuals with ASD and typically developed individuals (12), potential confounding effects of brain structural differences should be controlled for. Therefore, additional analyses were conducted controlling for potential confounders, including total intracranial volume and regional volume, where significant differences in MC-I availability were found between the ASD and typically developed participants, such as those seen in the ACC (see the Results section). Therefore, T1-weighted images were analyzed using the Computational Anatomy Toolbox (CAT12) (http://www.neuro.uni-jena.de/cat) for SPM12. First, individual T1-weighted images were segmented into gray matter, white matter, and CSF via the standard segmentation function implemented in CAT12. In this process, individual total intracranial volume was automatically estimated. Regional gray matter volume in the ACC ROI was calculated by first using CAT12 to normalize the segmented gray matter images and individual ACC ROI images to Montreal Neurological Institute space, and then using the get_totals MATLAB script (http://www0.cs.ucl.ac.uk/staff/g.ridgway/vbm/get_totals.m) to calculate regional gray matter volume within the normalized ACC mask images.
ROI Analysis
We set the ACC, superior temporal gyrus, occipital cortex, dorsolateral prefrontal cortex, thalamus, and primary motor cortex as the ROIs based on previous reports of mitochondrial abnormalities from postmortem studies of ASD (see Figure S1 in the online supplement ) (5, 6, 8, 13). The ROIs in individual MRI images were traced manually using PMOD. Although ROIs should be defined separately for each individual before spatial normalization, it has been suggested that warping of the atlases in the transformation process of individual PET images by the automated creation of ROIs can lead to mis-delineation (14). Conversely, hand-drawn ROI delineation is more exact but relies on subjective choices. To minimize any intersubject variance resulting from operator-dependent choices, a single blinded operator (Ya.K.) delineated all the ROIs for all participants in this study. We then transferred each ROI onto the corresponding SUVR parametric images to obtain the SUVR for each ROI. Our ROI approach on MRI followed by the placement of ROIs on high-spatial-resolution PET images (2.4 mm full width at half maximum) allowed us to exclude a significant partial volume effect problem.
Statistical Analysis
During the design of the study, because no previous [18F]BCPP-EF PET study testing psychiatric disorders was available, predicting effect size was difficult. Based on previous PET studies comparing participants with ASD and typically developed individuals and reporting significant diagnostic differences (15–17), we expected a target sample size of 20 in each group to be adequate, and we set the required number of participants in each group at a maximum of 30, with an assumed withdrawal rate less than 50%. After registration of 20 participants in each group to be analyzed, we decided to complete recruitment at 24 participants in each group, with withdrawal of just one participant in the ASD group (see the Results section).
All statistical analyses were conducted using Stata, version 15.0 (StataCorp, College Station, Tex.). For the demographic variables, categorical variables were assessed using the chi-square test, and continuous variables were assessed using the independent-samples t test. The threshold for statistical significance was set at a p value of <0.05. To compare the SUVR of individuals with ASD with that of the typically developed participants and test regional specificity of the difference in SUVR, we used analysis of variance (ANOVA), with group as the between-subject factor and ROI as the within-subject factor. In the presence of significant main interactions between group and ROI, regional contrasts in the six ROIs were examined using post hoc t tests with the Bonferroni correction for multiple comparisons, with a p value threshold of <0.0083 (0.05/6 ROIs).
To identify the clinical correlates of the identified aberrant SUVR in participants with ASD, the relationships between ADOS-2 subscales and the SUVR in brain regions with significant differences between the ASD and typically developed groups were examined using Pearson’s correlation analysis in participants with ASD. The Bonferroni-corrected significance threshold was set at a p value of <0.0167 (0.05/3 subscales×1 ROI).
Results
Participants
Twenty-four males with ASD and 24 typically developed males were recruited from October 31, 2017, to April 1, 2020. Of the 48 participants in the study, one participant with ASD was excluded. Because he was experiencing pain in his neck during the PET scan and was not able to remain motionless, we stopped scanning in the middle of the scan at the participant’s request. None of the participants had any current neuropsychiatric comorbidities, clinical manifestations of mitochondrial diseases, or abnormal plasma lactate or pyruvate levels—indicating the existence of mitochondrial diseases—based on a definition in previous studies (plasma lactate level >2 mM, pyruvate level >0.13 mM, and lactate/pyruvate ratio >22 or <8) (3, 4, 18). There were no significant differences between the ASD and typically developed groups in age, height, body weight, parental socioeconomic status, or full-scale IQ (Table 1). Individuals with ASD had significantly higher total Autism Spectrum Quotient scores and significantly lower socioeconomic status than the typically developed participants. Although no participant had a diagnosis of depression or an anxiety disorder, those in the ASD group showed significantly higher scores on the CES-D and the STAI state and trait scales compared with the typically developed group.
| Participants With ASD (N=23) | Typically Developed Participants (N=24) | Analysis | ||||
|---|---|---|---|---|---|---|
| Variable | Mean | SD | Mean | SD | t | p |
| Ageb (years) | 25.3 | 6.4 | 24.4 | 6.1 | 0.53 | 0.600 |
| Height (cm) | 168.7 | 6.0 | 171.3 | 4.9 | 1.61 | 0.114 |
| Body weight (kg) | 68.1 | 16.4 | 63.2 | 8.3 | 1.33 | 0.191 |
| Socioeconomic statusc | 3.5 | 1.1 | 2.4 | 0.6 | 4.43 | 0.001 |
| Parental socioeconomic statusc | 2.6 | 1.0 | 2.5 | 0.8 | 0.26 | 0.797 |
| IQ | ||||||
| Full-scale IQ | 104.8 | 16.3 | 106.6 | 14.8 | 0.40 | 0.694 |
| Verbal IQ | 108.4 | 16.0 | 108.0 | 14.6 | 0.10 | 0.923 |
| Performance IQ | 99.5 | 16.9 | 103.3 | 14.9 | 0.81 | 0.422 |
| Autism Spectrum Quotient | 30.5 | 7.2 | 17.2 | 7.2 | 6.35 | <0.001 |
| ADI-R | ||||||
| Reciprocity | 17.9 | 8.0 | ||||
| Communication | 12.9 | 5.4 | ||||
| Repetitive | 4.3 | 2.9 | ||||
| ADOS-2 | ||||||
| Reciprocity | 3.2 | 1.4 | ||||
| Communication | 7.1 | 1.8 | ||||
| Repetitive behavior | 1.5 | 1.3 | ||||
| CES-D | 17.0 | 9.8 | 9.0 | 5.5 | 3.47 | 0.001 |
| STAI | ||||||
| State score | 48.0 | 12.4 | 38.0 | 6.4 | 3.52 | 0.001 |
| Trait score | 51.3 | 13.4 | 40.8 | 7.4 | 3.33 | 0.002 |
a
ADI-R=Autism Diagnostic Interview–Revised; ADOS-2=Autism Diagnostic Observation Schedule, 2nd ed.; ASD=autism spectrum disorder; CES-D=Center for Epidemiologic Studies Depression Scale; STAI=State-Trait Anxiety Inventory.
b
The age range was 18–44 years in the ASD group and 18–42 years in the typically developed group.
c
Assessed by the Hollingshead index; a higher score indicates a lower socioeconomic status.
Group Differences in the SUVR of [18F]BCPP-EF
Averaged and representative [18F]BCPP-EF PET images are shown in Figure 1 and in Figure S2 in the online supplement . In the [18F]BCPP-EF PET measurements, there was a significant interaction between group and ROI (F=7.80, df=5, 41, p<0.001) and a significant main effect of ROI (F=70.09, df=5, 41, p<0.001), while the main effect of group was not significant (F=3.18, df=1, 45, p=0.081) in ANOVA. Post hoc t tests for the significant interaction between group status and ROI indicated that the SUVR of [18F]BCPP-EF in the ACC was significantly lower in the ASD group than the typically developed group (t=3.37, df=1, 45, Bonferroni-corrected p=0.012; Cohen’s d=0.98) (Table 2 and Figure 2), whereas there were no significant diagnostic differences in the other regions. The additional analyses confirmed that the statistical conclusions were preserved after considering potential confounding effects of intracranial volume, ACC volume, and subthreshold depression or anxiety. There was no significant correlation of ACC SUVR with CES-D or state-trait STAI scores in the typically developed group or the ASD group (for details, see the online supplement ).
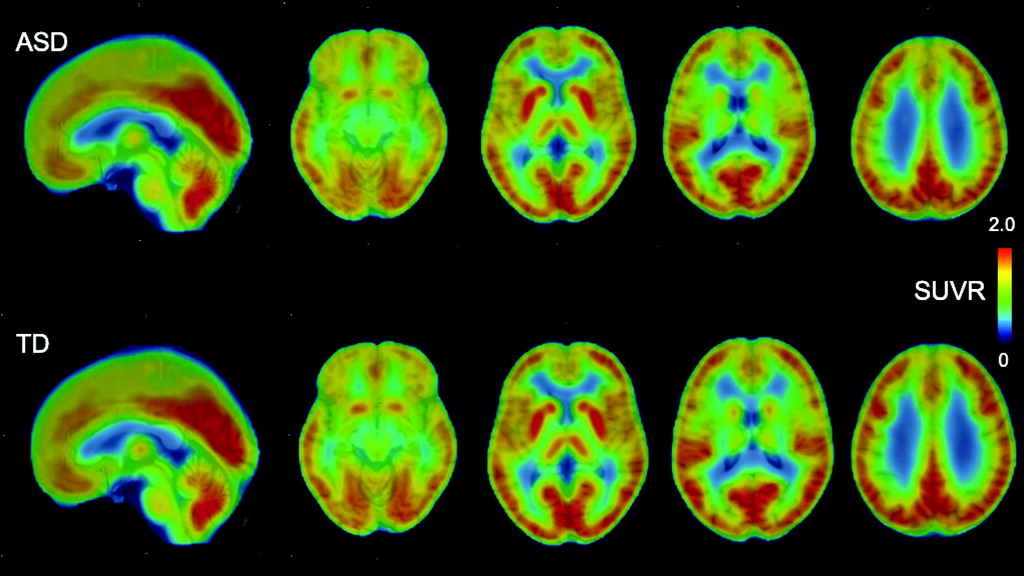
FIGURE 1. Averaged PET images of SUVR of [18F]BCPP-EF in participants with ASD and typically developed participantsa
aAfter coregistration with the individual T1-weighted images and normalization to Montreal Neurological Institute space using SPM12, individual SUVR images were averaged across participants in each group to obtain mean parametric [18F]BCPP-EF SUVR images. ASD=autism spectrum disorder; SUVR=standard uptake value ratio; TD=typically developed.
| Participants With ASD (N=23) | Typically Developed Participants (N=24) | Analysis | |||||
|---|---|---|---|---|---|---|---|
| Region of Interest | Mean | SD | Mean | SD | t | p | Cohen’s d |
| Anterior cingulate cortex | 0.77 | 0.12 | 0.87 | 0.07 | 3.37 | 0.0016b | 0.98 |
| Thalamus | 0.99 | 0.13 | 1.04 | 0.13 | 1.36 | 0.182 | 0.40 |
| Superior temporal gyrus | 0.83 | 0.08 | 0.85 | 0.05 | 0.98 | 0.333 | 0.29 |
| Occipital cortex | 1.03 | 0.17 | 1.11 | 0.10 | 1.89 | 0.065 | 0.55 |
| Dorsolateral prefrontal cortex | 0.94 | 0.13 | 0.92 | 0.08 | 0.62 | 0.538 | 0.18 |
| Primary motor cortex | 0.90 | 0.11 | 0.89 | 0.07 | 0.59 | 0.563 | 0.17 |
a
ASD=autism spectrum disorder; SUVR=standard uptake value ratio.
b
Statistically significant after Bonferroni correction.
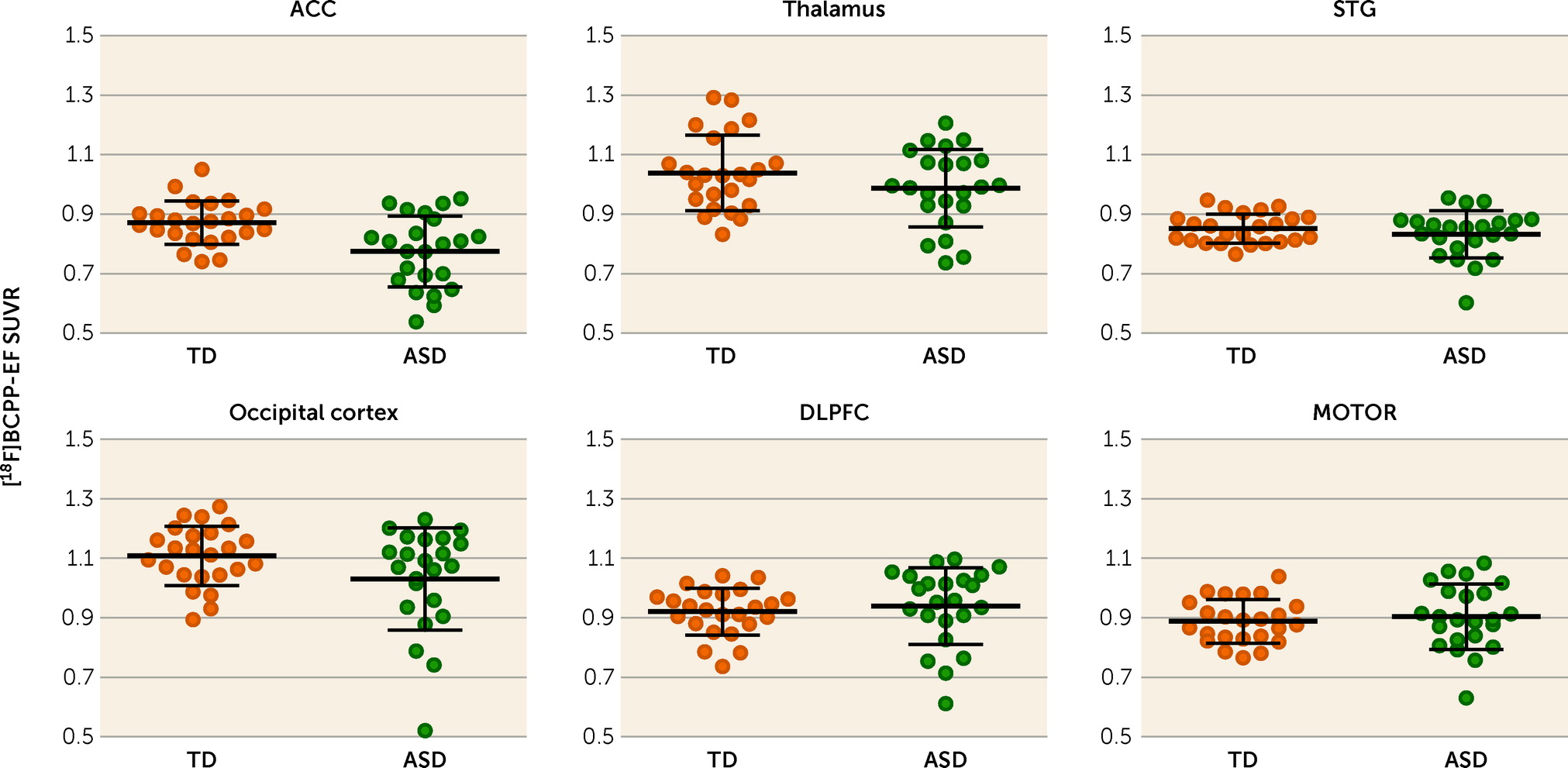
FIGURE 2. Differences in [18F]BCPP-EF SUVR in the regions of interest between participants with ASD and typically developed participantsa
aPlots represent the regional [18F]BCPP-EF SUVR in the regions of interest in the two groups. Mean values are indicated by horizontal bars, and error bars indicate standard deviation. As indicated by the asterisk, there was a significant group difference in the ACC (t=3.37, uncorrected p=0.002, Bonferroni-corrected p=0.012). ACC=anterior cingulate cortex; ASD=autism spectrum disorder; DLPFC=dorsolateral prefrontal cortex; MOTOR=primary motor cortex; STG=superior temporal gyrus; SUVR=standard uptake value ratio; TD=typically developed.
Correlations Between the SUVR of [18F]BCPP-EF and ASD Core Symptoms
One individual with ASD was excluded from the correlational analyses with ADOS-2 scores because the ADOS-2 could not be administered. After the exclusion of this participant, the significantly lower SUVR of [18F]BCPP-EF in the ACC of participants with ASD (N=22) than typically developed participants (N=24) was preserved (t=3.66, df=44, Bonferroni-corrected p=0.008) (see Table S1 in the online supplement ).
The analysis showed a significant negative correlation between the ADOS-2 communication score and the [18F]BCPP-EF SUVR in the ACC (r=−0.54, Bonferroni-corrected p=0.0297) (Figure 3A; see also Table S2 in the online supplement ). The reciprocity subscale and the restricted and repetitive behaviors subscale were not significantly correlated with the SUVR in the ACC (Figure 3B,C; see also Table S2 in the online supplement ).
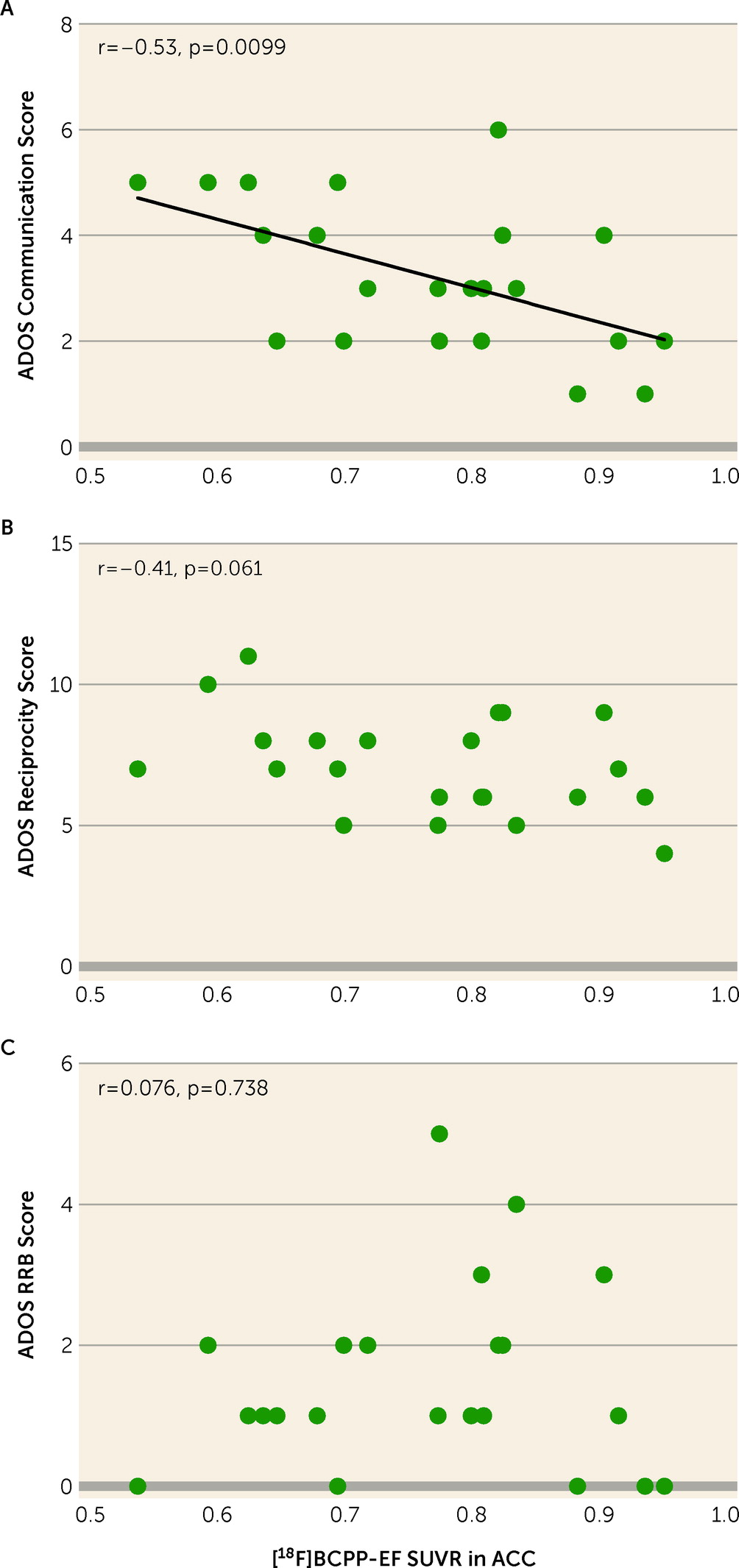
FIGURE 3. Correlations between ADOS-2 subscales and [18F]BCPP-EF SUVR of the ACC in participants with ASDa
aScatterplots show correlations between scores on the ADOS-2 communication subscale, reciprocity subscale, and restricted and repetitive behaviors subscale and the [18F]BCPP-EF SUVR of the region of interest that had a significant group difference between the ASD and typically developed groups in participants with ASD (N=22), namely, the ACC. ACC=anterior cingulate cortex; ADOS-2=Autism Diagnostic Observation Schedule, 2nd ed.; ASD=autism spectrum disorder; RRB=restricted and repetitive behaviors; SUVR=standard uptake value ratio.
Discussion
To the best of our knowledge, the present study demonstrates for the first time lower MC-I availability in the ACC of psychotropic medication–free and mitochondrial disease–free adult males with high-functioning ASD compared with demographically matched typically developed individuals. The decreased availability of MC-I in the ACC of participants with ASD was significantly correlated with the severe ADOS-2 communication score. As revealed in our previous studies, the decreased [18F]BCPP-EF binding indicates brain regional mitochondrial dysfunction, including the degradation and quantitative reduction of MC-I proteins as well as reduced functional activities of MC-I (for details, see the online supplement ).
The low availability of MC-I in individuals with ASD compared with typically developed individuals is consistent with previous reports suggesting a critical role of mitochondrial dysfunction in the pathogenesis of ASD. A meta-analysis of peripheral biomarkers (4) showed the high prevalence of mitochondrial disease and abnormal values in lactate, pyruvate, alanine, creatine kinase, ammonia, and aspartate aminotransferase in the general population of individuals with ASD. Reduced mitochondrial complex activities and protein levels in postmortem brain (especially in the frontal cortices, including the ACC) and in peripheral samples have repeatedly been reported in conjunction with alterations in MC-I in individuals with ASD (3, 4, 18). A downregulation in MC-I genes in the postmortem autistic brain and associations between mitochondrial DNA variants and ASD have also been reported (3, 5, 19). The elevated production of reactive oxygen species, which are produced during the metabolism of oxygen at the electron transport chain including MC-I, and increased markers of oxidative stress were reported in individuals with ASD in another meta-analysis (20). Moreover, magnetic resonance spectroscopy studies investigating brain lactate have suggested abnormalities in mitochondrial energy metabolism (21, 22). In addition, previous postmortem studies as well as in vivo PET studies have reported the abnormal expression of translocator protein, which is an activated glial marker that is expressed on mitochondrial external membranes, and have suggested mitochondrial dysfunction in the autistic brain (16, 23, 24). Translocator protein has been implicated in several physiological processes, including immune modulation and mitochondrial homeostasis, while BCPP specifically binds to MC-I and reflects mitochondrial function regardless of immune modulation. Our results support these previous studies implicating mitochondrial dysfunction, and specifically lower MC-I, in the pathophysiology of ASD, and further extend the evidence for mitochondrial dysfunction in ASD by revealing lower levels of MC-I in the living brain for the first time.
In line with the present finding of decreased MC-I availability in the ACC of individuals with ASD, previous studies have supported an important role of the ACC in the pathophysiology of ASD. Functional MRI studies have repeatedly revealed dysfunction of the ACC in individuals with ASD (25). Additionally, a lower density of myelinated axons and a decreased expression of axon guidance receptors have been identified in postmortem brains of individuals with ASD (26). Because mitochondria regulate axon guidance receptors, decreased MC-I activity may lead to malfunctions in axonal outgrowth (27), and the ensuing disorganization in axon guidance may disrupt functional and structural connectivity in individuals with ASD (28). The present study further offers direct evidence for lower MC-I activity in the ACC in vivo in individuals with ASD.
Although previous postmortem studies have reported decreased nuclear gene expression of MC-I in all of the ROIs that we investigated, our study detected a significant group difference in MC-I availability specifically in the ACC. This discrepancy might be explained by the potential confounding factors of postmortem studies. In postmortem studies, the major causes of death in individuals with ASD were drowning, asphyxiation, and seizure, which led to a hypoxic event (5, 6, 13). Hypoxia is reported to deactivate the MC-I (29), while seizure activity impairs mitochondrial energy production by affecting the activity of mitochondrial enzymes (30). Furthermore, previous reports have suggested that antidepressants, antipsychotics, and selective serotonin reuptake inhibitors impair MC-I activity and ATP production (31, 32). All of these factors might therefore affect the results of postmortem studies (5, 6, 13). In contrast, because no participant in the present study was taking psychotropic medication or had a seizure history, it is unlikely that these factors confounded the study results.
The significant correlation between reduced MC-I activity in ACC and high ADOS-2 communication scores suggests that mitochondrial dysfunction in ACC plays an important role in ASD social communication deficits. Previous neuroimaging studies have also supported a significant contribution of the ACC to ASD social communication deficits. Based on hypoactivation of the ACC in response to social rewards in ASD (25), the social motivation theory suggests that individuals with ASD experience less reward for social interactions than do typically developed individuals (33). Reduced glucose metabolism in the ACC, as assessed by 18F-FDG PET, which is a well-established technique for the quantitative measurement of the regional metabolic rate of glucose in the brain and reflects glucose metabolism as well as neuroinflammation, has also been reported to correlate with more severe ADI-R social domain deficits (34). PET imaging with 18F-BCPP-EF, which has specificity for MC-I activity and which is not affected by inflammation with microglial activation, could be expected to provide more accurate information about neurodevelopmental pathophysiology than 18F-FDG. Moreover, levels of N-acetylaspartate, which is synthesized in mitochondria and reflects MC-I activity, are correlated with ADOS-2 social affect scores in individuals with ASD (35). In the ACC of postmortem ASD brains, the dysregulation of excitatory neuron-related genes was reported to correlate with total ADI-R score (36). Our results in the present study further suggest that MC-I dysfunction is a candidate mechanism underlying the role of the ACC in autistic social communication deficits.
The finding of MC-I dysfunction in the present study signals its possible importance as a novel therapeutic target for ASD. A randomized trial showed that folinic acid, which is one of the vitamins that are needed for MC-I activity (37), improves verbal communication in children with ASD (38). Furthermore, several studies have suggested that the ACC, where we found mitochondrial dysfunction, is associated with the neural processes underlying therapeutic mechanisms in ASD, such as improved social behaviors with genetic activation of SHANK3 in a mouse model having dysfunctional pyramidal neurons in the ACC (39), improved activity and functional connectivity in the ACC with oxytocin administration (40, 41), and modified neural plasticity associated with the N-methyl-d-aspartate receptor (NMDAR) induced by NMDAR agonists or oxytocin (42, 43). The present study provides further evidence to support the contribution of MC-I dysfunction in the ACC to the core symptoms of ASD, thus indicating its potential as a novel therapeutic target.
Some methodological considerations and limitations of the present study must be borne in mind. First, the participants were restricted to high-functioning young adult Japanese males, free of psychotropic medication. Although the uniformity of demographic characteristics enhanced our ability to detect the difference in MC-I availability in the ACC of ASD participants and typically developed participants with a large effect size (d=0.98), care should be taken when generalizing the study findings. The effect of MC-I availability might be found to be smaller in other ASD populations, such as females, medicated individuals, and children. Second, although the target sample size of our study was determined on the basis of previous PET studies in ASD (15–17), a future study with a larger sample size might detect significant associations between MC-I availability and other core symptoms of ASD that had marginal significance in the present sample (for example, the association with ADOS-2 reciprocity score approached significance at p=0.061; see Table S2 in the online supplement ). Third, our study was a cross-sectional comparison, and we are therefore unable to infer causality for the role of MC-I availability in the pathophysiology of ASD. Fourth, this study did not evaluate regional cerebral blood flow. Although previous studies have shown that [18F]BCPP-EF availability was not influenced by regional cerebral blood flow in the conscious monkey brain (44, 45), the possibility that regional blood flow affects [18F]BCPP-EF availability in the human brain cannot be ruled out. Fifth, given the difficulty associated with the placement of an invasive arterial line that would allow assessment of volumes of distribution in participants with ASD, in the present study, we utilized the surrogate SUVR measure instead of volumes of distribution. Although our previous studies have shown correlations between volumes of distribution and SUVR, these were not in a disease-specific manner (46, 47), which casts doubt on whether we could make any inferences regarding high-functioning ASD. Sixth, to minimize potential confounding effects, we recruited participants who were not taking any psychotropic medication and did not have any psychiatric comorbidities, and we screened their peripheral blood to rule out mitochondrial diseases. Even with these steps, we cannot rule out medical comorbidities that might increase the chances of finding mitochondrial dysfunction.
In conclusion, this study provides the first evidence from in vivo human brain of lower MC-I binding in the ACC of individuals with ASD compared with typically developed individuals, and of its relationship to severe social communication deficits. These findings indicate an important role of MC-I in the pathophysiology of ASD and suggest the potential of MC-I as a novel molecular target for pharmacological treatments of ASD core symptoms.
Acknowledgments
The authors express their gratitude to the study participants and to the staff of the Hamamatsu Medical Imaging Center, the Hamamatsu Medical Photonics Foundation, the Global Strategic Challenge Center, Hamamatsu Photonics K.K., and the Department of Psychiatry, Hamamatsu University School of Medicine, for their assistance with data collection, and especially Rie Gonda, Emiko Hatano, and Miyuki Suzuki for their efforts on data management. The authors also thank Bronwen Gardner, Ph.D., from Edanz, for editing a draft of this manuscript.
Supplementary Material
File (appi.ajp.22010014.ds001.pdf)
- View/Download
- 426.51 KB
References
1.
Maenner MJ, Shaw KA, Bakian AV, et al: Prevalence and characteristics of autism spectrum disorder among children aged 8 years: autism and developmental disabilities monitoring network, 11 sites, United States, 2018. MMWR Surveill Summ 2021; 70:1–16
2.
American Psychiatric Association: Diagnostic and Statistical Manual of Mental Disorders, 5th ed. Washington, DC, American Psychiatric Association, 2013
3.
Hollis F, Kanellopoulos AK, Bagni C: Mitochondrial dysfunction in autism spectrum disorder: clinical features and perspectives. Curr Opin Neurobiol 2017; 45:178–187
4.
Rossignol DA, Frye RE: Mitochondrial dysfunction in autism spectrum disorders: a systematic review and meta-analysis. Mol Psychiatry 2012; 17:290–314
5.
Anitha A, Nakamura K, Thanseem I, et al: Downregulation of the expression of mitochondrial electron transport complex genes in autism brains. Brain Pathol 2013; 23:294–302
6.
Chauhan A, Gu F, Essa MM, et al: Brain region–specific deficit in mitochondrial electron transport chain complexes in children with autism. J Neurochem 2011; 117:209–220
7.
Harris JJ, Jolivet R, Attwell D: Synaptic energy use and supply. Neuron 2012; 75:762–777
8.
Tang G, Gutierrez Rios P, Kuo SH, et al: Mitochondrial abnormalities in temporal lobe of autistic brain. Neurobiol Dis 2013; 54:349–361
9.
Harada N, Nishiyama S, Kanazawa M, et al: Development of novel PET probes, [18F]BCPP-EF, [18F]BCPP-BF, and [11C]BCPP-EM for mitochondrial complex 1 imaging in the living brain. J Labelled Comp Radiopharm 2013; 56:553–561
10.
Terada T, Obi T, Bunai T, et al: In vivo mitochondrial and glycolytic impairments in patients with Alzheimer disease. Neurology 2020; 94:e1592–e1604
11.
Ouchi Y, Nobezawa S, Okada H, et al: Altered glucose metabolism in the hippocampal head in memory impairment. Neurology 1998; 51:136–142
12.
Lukito S, Norman L, Carlisi C, et al: Comparative meta-analyses of brain structural and functional abnormalities during cognitive control in attention-deficit/hyperactivity disorder and autism spectrum disorder. Psychol Med 2020; 50:894–919
13.
Gu F, Chauhan V, Kaur K, et al: Alterations in mitochondrial DNA copy number and the activities of electron transport chain complexes and pyruvate dehydrogenase in the frontal cortex from subjects with autism. Transl Psychiatry 2013; 3:e299
14.
Karjalainen T, Tuisku J, Santavirta S, et al: Magia: robust automated image processing and kinetic modeling toolbox for PET neuroinformatics. Front Neuroinform 2020; 14:3
15.
Nakamura K, Sekine Y, Ouchi Y, et al: Brain serotonin and dopamine transporter bindings in adults with high-functioning autism. Arch Gen Psychiatry 2010; 67:59–68
16.
Suzuki K, Sugihara G, Ouchi Y, et al: Microglial activation in young adults with autism spectrum disorder. JAMA Psychiatry 2013; 70:49–58
17.
Suzuki K, Sugihara G, Ouchi Y, et al: Reduced acetylcholinesterase activity in the fusiform gyrus in adults with autism spectrum disorders. Arch Gen Psychiatry 2011; 68:306–313
18.
Giulivi C, Zhang YF, Omanska-Klusek A, et al: Mitochondrial dysfunction in autism. JAMA 2010; 304:2389–2396
19.
Chalkia D, Singh LN, Leipzig J, et al: Association between mitochondrial DNA haplogroup variation and autism spectrum disorders. JAMA Psychiatry 2017; 74:1161–1168
20.
Chen L, Shi XJ, Liu H, et al: Oxidative stress marker aberrations in children with autism spectrum disorder: a systematic review and meta-analysis of 87 studies (N=9109). Transl Psychiatry 2021; 11:15
21.
Minshew NJ, Goldstein G, Dombrowski SM, et al: A preliminary 31P MRS study of autism: evidence for undersynthesis and increased degradation of brain membranes. Biol Psychiatry 1993; 33:762–773
22.
Goh S, Dong Z, Zhang Y, et al: Mitochondrial dysfunction as a neurobiological subtype of autism spectrum disorder: evidence from brain imaging. JAMA Psychiatry 2014; 71:665–671
23.
Anitha A, Nakamura K, Thanseem I, et al: Brain region-specific altered expression and association of mitochondria-related genes in autism. Mol Autism 2012; 3:12
24.
Zurcher NR, Loggia ML, Mullett JE, et al: [11C]PBR28 MR-PET imaging reveals lower regional brain expression of translocator protein (TSPO) in young adult males with autism spectrum disorder. Mol Psychiatry 2021; 26:1659–1669
25.
Clements CC, Zoltowski AR, Yankowitz LD, et al: Evaluation of the social motivation hypothesis of autism: a systematic review and meta-analysis. JAMA Psychiatry 2018; 75:797–808
26.
Suda S, Iwata K, Shimmura C, et al: Decreased expression of axon-guidance receptors in the anterior cingulate cortex in autism. Mol Autism 2011; 2:14
27.
Smith GM, Gallo G: The role of mitochondria in axon development and regeneration. Dev Neurobiol 2018; 78:221–237
28.
Zikopoulos B, Barbas H: Changes in prefrontal axons may disrupt the network in autism. J Neurosci 2010; 30:14595–14609
29.
Hernansanz-Agustin P, Ramos E, Navarro E, et al: Mitochondrial complex I deactivation is related to superoxide production in acute hypoxia. Redox Biol 2017; 12:1040–1051
30.
Liang LP, Patel M: Seizure-induced changes in mitochondrial redox status. Free Radic Biol Med 2006; 40:316–322
31.
Rosenfeld M, Brenner-Lavie H, Ari SG-B, et al: Perturbation in mitochondrial network dynamics and in complex I dependent cellular respiration in schizophrenia. Biol Psychiatry 2011; 69:980–988
32.
Weinbach EC, Costa JL, Nelson BD, et al: Effects of tricyclic antidepressant drugs on energy-linked reactions in mitochondria. Biochem Pharmacol 1986; 35:1445–1451
33.
Chevallier C, Kohls G, Troiani V, et al: The social motivation theory of autism. Trends Cogn Sci 2012; 16:231–239
34.
Haznedar MM, Buchsbaum MS, Wei TC, et al: Limbic circuitry in patients with autism spectrum disorders studied with positron emission tomography and magnetic resonance imaging. Am J Psychiatry 2000; 157:1994–2001
35.
Peterson BS, Zargarian A, Peterson JB, et al: Hyperperfusion of frontal white and subcortical gray matter in autism spectrum disorder. Biol Psychiatry 2019; 85:584–595
36.
Velmeshev D, Schirmer L, Jung D, et al: Single-cell genomics identifies cell type-specific molecular changes in autism. Science 2019; 364:685–689
37.
Morscher RJ, Ducker GS, Li SH, et al: Mitochondrial translation requires folate-dependent tRNA methylation. Nature 2018; 554:128–132
38.
Frye RE, Slattery J, Delhey L, et al: Folinic acid improves verbal communication in children with autism and language impairment: a randomized double-blind placebo-controlled trial. Mol Psychiatry 2018; 23:247–256
39.
Guo B, Chen J, Chen Q, et al: Anterior cingulate cortex dysfunction underlies social deficits in Shank3 mutant mice. Nat Neurosci 2019; 22:1223–1234
40.
Watanabe T, Abe O, Kuwabara H, et al: Mitigation of sociocommunicational deficits of autism through oxytocin-induced recovery of medial prefrontal activity: a randomized trial. JAMA Psychiatry 2014; 71:166–175
41.
Watanabe T, Kuroda M, Kuwabara H, et al: Clinical and neural effects of six-week administration of oxytocin on core symptoms of autism. Brain 2015; 138:3400–3412
42.
Kato Y, Kuwabara H, Okada T, et al: Oxytocin-induced increase in N,N-dimethylglycine and time course of changes in oxytocin efficacy for autism social core symptoms. Mol Autism 2021; 12:15
43.
Benner S, Aoki Y, Watanabe T, et al: Neurochemical evidence for differential effects of acute and repeated oxytocin administration. Mol Psychiatry 2021; 26:710–720
44.
Fang J, Ohba H, Hashimoto F, et al: Imaging mitochondrial complex I activation during a vibrotactile stimulation: a PET study using [18F]BCPP-EF in the conscious monkey brain. J Cereb Blood Flow Metab 2020; 40:2521–2532
45.
Tsukada H, Ohba H, Nishiyama S, et al: PET imaging of ischemia-induced impairment of mitochondrial complex I function in monkey brain. J Cereb Blood Flow Metab 2014; 34:708–714
46.
Terada T, Therriault J, Kang MSP, et al: Mitochondrial complex I abnormalities is associated with tau and clinical symptoms in mild Alzheimer’s disease. Mol Neurodegener 2021; 16:28
47.
Mansur A, Rabiner EA, Tsukada H, et al: Test-retest variability and reference region-based quantification of 18F-BCPP-EF for imaging mitochondrial complex I in the human brain. J Cereb Blood Flow Metab 2021; 41:771–779
Information & Authors
Information
Published In


History
Received: 6 January 2022
Revision received: 21 March 2022
Revision received: 21 April 2022
Accepted: 2 May 2022
Published online: 7 September 2022
Published in print: April 01, 2023
Keywords
Authors
Competing Interests
The authors report no financial relationships with commercial interests.
Funding Information
Supported by the Strategic Research Program for Brain Sciences of the Japan Agency for Medical Research and Development under grant JP16dm0107134.
Metrics & Citations
Metrics
Citations
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
For more information or tips please see 'Downloading to a citation manager' in the Help menu.
View Options
View options
PDF/EPUB
View PDF/EPUBFigures
FIGURE 1. Averaged PET images of SUVR of [18F]BCPP-EF in participants with ASD and typically developed participantsa
aAfter coregistration with the individual T1-weighted images and normalization to Montreal Neurological Institute space using SPM12, individual SUVR images were averaged across participants in each group to obtain mean parametric [18F]BCPP-EF SUVR images. ASD=autism spectrum disorder; SUVR=standard uptake value ratio; TD=typically developed.
FIGURE 2. Differences in [18F]BCPP-EF SUVR in the regions of interest between participants with ASD and typically developed participantsa
aPlots represent the regional [18F]BCPP-EF SUVR in the regions of interest in the two groups. Mean values are indicated by horizontal bars, and error bars indicate standard deviation. As indicated by the asterisk, there was a significant group difference in the ACC (t=3.37, uncorrected p=0.002, Bonferroni-corrected p=0.012). ACC=anterior cingulate cortex; ASD=autism spectrum disorder; DLPFC=dorsolateral prefrontal cortex; MOTOR=primary motor cortex; STG=superior temporal gyrus; SUVR=standard uptake value ratio; TD=typically developed.
FIGURE 3. Correlations between ADOS-2 subscales and [18F]BCPP-EF SUVR of the ACC in participants with ASDa
aScatterplots show correlations between scores on the ADOS-2 communication subscale, reciprocity subscale, and restricted and repetitive behaviors subscale and the [18F]BCPP-EF SUVR of the region of interest that had a significant group difference between the ASD and typically developed groups in participants with ASD (N=22), namely, the ACC. ACC=anterior cingulate cortex; ADOS-2=Autism Diagnostic Observation Schedule, 2nd ed.; ASD=autism spectrum disorder; RRB=restricted and repetitive behaviors; SUVR=standard uptake value ratio.
Tables
Media
References
References
1.
Maenner MJ, Shaw KA, Bakian AV, et al: Prevalence and characteristics of autism spectrum disorder among children aged 8 years: autism and developmental disabilities monitoring network, 11 sites, United States, 2018. MMWR Surveill Summ 2021; 70:1–16
2.
American Psychiatric Association: Diagnostic and Statistical Manual of Mental Disorders, 5th ed. Washington, DC, American Psychiatric Association, 2013
3.
Hollis F, Kanellopoulos AK, Bagni C: Mitochondrial dysfunction in autism spectrum disorder: clinical features and perspectives. Curr Opin Neurobiol 2017; 45:178–187
4.
Rossignol DA, Frye RE: Mitochondrial dysfunction in autism spectrum disorders: a systematic review and meta-analysis. Mol Psychiatry 2012; 17:290–314
5.
Anitha A, Nakamura K, Thanseem I, et al: Downregulation of the expression of mitochondrial electron transport complex genes in autism brains. Brain Pathol 2013; 23:294–302
6.
Chauhan A, Gu F, Essa MM, et al: Brain region–specific deficit in mitochondrial electron transport chain complexes in children with autism. J Neurochem 2011; 117:209–220
7.
Harris JJ, Jolivet R, Attwell D: Synaptic energy use and supply. Neuron 2012; 75:762–777
8.
Tang G, Gutierrez Rios P, Kuo SH, et al: Mitochondrial abnormalities in temporal lobe of autistic brain. Neurobiol Dis 2013; 54:349–361
9.
Harada N, Nishiyama S, Kanazawa M, et al: Development of novel PET probes, [18F]BCPP-EF, [18F]BCPP-BF, and [11C]BCPP-EM for mitochondrial complex 1 imaging in the living brain. J Labelled Comp Radiopharm 2013; 56:553–561
10.
Terada T, Obi T, Bunai T, et al: In vivo mitochondrial and glycolytic impairments in patients with Alzheimer disease. Neurology 2020; 94:e1592–e1604
11.
Ouchi Y, Nobezawa S, Okada H, et al: Altered glucose metabolism in the hippocampal head in memory impairment. Neurology 1998; 51:136–142
12.
Lukito S, Norman L, Carlisi C, et al: Comparative meta-analyses of brain structural and functional abnormalities during cognitive control in attention-deficit/hyperactivity disorder and autism spectrum disorder. Psychol Med 2020; 50:894–919
13.
Gu F, Chauhan V, Kaur K, et al: Alterations in mitochondrial DNA copy number and the activities of electron transport chain complexes and pyruvate dehydrogenase in the frontal cortex from subjects with autism. Transl Psychiatry 2013; 3:e299
14.
Karjalainen T, Tuisku J, Santavirta S, et al: Magia: robust automated image processing and kinetic modeling toolbox for PET neuroinformatics. Front Neuroinform 2020; 14:3
15.
Nakamura K, Sekine Y, Ouchi Y, et al: Brain serotonin and dopamine transporter bindings in adults with high-functioning autism. Arch Gen Psychiatry 2010; 67:59–68
16.
Suzuki K, Sugihara G, Ouchi Y, et al: Microglial activation in young adults with autism spectrum disorder. JAMA Psychiatry 2013; 70:49–58
17.
Suzuki K, Sugihara G, Ouchi Y, et al: Reduced acetylcholinesterase activity in the fusiform gyrus in adults with autism spectrum disorders. Arch Gen Psychiatry 2011; 68:306–313
18.
Giulivi C, Zhang YF, Omanska-Klusek A, et al: Mitochondrial dysfunction in autism. JAMA 2010; 304:2389–2396
19.
Chalkia D, Singh LN, Leipzig J, et al: Association between mitochondrial DNA haplogroup variation and autism spectrum disorders. JAMA Psychiatry 2017; 74:1161–1168
20.
Chen L, Shi XJ, Liu H, et al: Oxidative stress marker aberrations in children with autism spectrum disorder: a systematic review and meta-analysis of 87 studies (N=9109). Transl Psychiatry 2021; 11:15
21.
Minshew NJ, Goldstein G, Dombrowski SM, et al: A preliminary 31P MRS study of autism: evidence for undersynthesis and increased degradation of brain membranes. Biol Psychiatry 1993; 33:762–773
22.
Goh S, Dong Z, Zhang Y, et al: Mitochondrial dysfunction as a neurobiological subtype of autism spectrum disorder: evidence from brain imaging. JAMA Psychiatry 2014; 71:665–671
23.
Anitha A, Nakamura K, Thanseem I, et al: Brain region-specific altered expression and association of mitochondria-related genes in autism. Mol Autism 2012; 3:12
24.
Zurcher NR, Loggia ML, Mullett JE, et al: [11C]PBR28 MR-PET imaging reveals lower regional brain expression of translocator protein (TSPO) in young adult males with autism spectrum disorder. Mol Psychiatry 2021; 26:1659–1669
25.
Clements CC, Zoltowski AR, Yankowitz LD, et al: Evaluation of the social motivation hypothesis of autism: a systematic review and meta-analysis. JAMA Psychiatry 2018; 75:797–808
26.
Suda S, Iwata K, Shimmura C, et al: Decreased expression of axon-guidance receptors in the anterior cingulate cortex in autism. Mol Autism 2011; 2:14
27.
Smith GM, Gallo G: The role of mitochondria in axon development and regeneration. Dev Neurobiol 2018; 78:221–237
28.
Zikopoulos B, Barbas H: Changes in prefrontal axons may disrupt the network in autism. J Neurosci 2010; 30:14595–14609
29.
Hernansanz-Agustin P, Ramos E, Navarro E, et al: Mitochondrial complex I deactivation is related to superoxide production in acute hypoxia. Redox Biol 2017; 12:1040–1051
30.
Liang LP, Patel M: Seizure-induced changes in mitochondrial redox status. Free Radic Biol Med 2006; 40:316–322
31.
Rosenfeld M, Brenner-Lavie H, Ari SG-B, et al: Perturbation in mitochondrial network dynamics and in complex I dependent cellular respiration in schizophrenia. Biol Psychiatry 2011; 69:980–988
32.
Weinbach EC, Costa JL, Nelson BD, et al: Effects of tricyclic antidepressant drugs on energy-linked reactions in mitochondria. Biochem Pharmacol 1986; 35:1445–1451
33.
Chevallier C, Kohls G, Troiani V, et al: The social motivation theory of autism. Trends Cogn Sci 2012; 16:231–239
34.
Haznedar MM, Buchsbaum MS, Wei TC, et al: Limbic circuitry in patients with autism spectrum disorders studied with positron emission tomography and magnetic resonance imaging. Am J Psychiatry 2000; 157:1994–2001
35.
Peterson BS, Zargarian A, Peterson JB, et al: Hyperperfusion of frontal white and subcortical gray matter in autism spectrum disorder. Biol Psychiatry 2019; 85:584–595
36.
Velmeshev D, Schirmer L, Jung D, et al: Single-cell genomics identifies cell type-specific molecular changes in autism. Science 2019; 364:685–689
37.
Morscher RJ, Ducker GS, Li SH, et al: Mitochondrial translation requires folate-dependent tRNA methylation. Nature 2018; 554:128–132
38.
Frye RE, Slattery J, Delhey L, et al: Folinic acid improves verbal communication in children with autism and language impairment: a randomized double-blind placebo-controlled trial. Mol Psychiatry 2018; 23:247–256
39.
Guo B, Chen J, Chen Q, et al: Anterior cingulate cortex dysfunction underlies social deficits in Shank3 mutant mice. Nat Neurosci 2019; 22:1223–1234
40.
Watanabe T, Abe O, Kuwabara H, et al: Mitigation of sociocommunicational deficits of autism through oxytocin-induced recovery of medial prefrontal activity: a randomized trial. JAMA Psychiatry 2014; 71:166–175
41.
Watanabe T, Kuroda M, Kuwabara H, et al: Clinical and neural effects of six-week administration of oxytocin on core symptoms of autism. Brain 2015; 138:3400–3412
42.
Kato Y, Kuwabara H, Okada T, et al: Oxytocin-induced increase in N,N-dimethylglycine and time course of changes in oxytocin efficacy for autism social core symptoms. Mol Autism 2021; 12:15
43.
Benner S, Aoki Y, Watanabe T, et al: Neurochemical evidence for differential effects of acute and repeated oxytocin administration. Mol Psychiatry 2021; 26:710–720
44.
Fang J, Ohba H, Hashimoto F, et al: Imaging mitochondrial complex I activation during a vibrotactile stimulation: a PET study using [18F]BCPP-EF in the conscious monkey brain. J Cereb Blood Flow Metab 2020; 40:2521–2532
45.
Tsukada H, Ohba H, Nishiyama S, et al: PET imaging of ischemia-induced impairment of mitochondrial complex I function in monkey brain. J Cereb Blood Flow Metab 2014; 34:708–714
46.
Terada T, Therriault J, Kang MSP, et al: Mitochondrial complex I abnormalities is associated with tau and clinical symptoms in mild Alzheimer’s disease. Mol Neurodegener 2021; 16:28
47.
Mansur A, Rabiner EA, Tsukada H, et al: Test-retest variability and reference region-based quantification of 18F-BCPP-EF for imaging mitochondrial complex I in the human brain. J Cereb Blood Flow Metab 2021; 41:771–779