Depression affects one in eight persons in the United States (
1) and is projected to become the second leading cause of disability in the world by the year 2020 (
2). However, generalizable evidence from clinical trials to inform treatment selection and sequencing is quite limited. Most clinical trial participants are recruited by advertisement rather than from representative practice settings. Eligibility criteria often exclude persons who have coexisting general medical or psychiatric disorders or who are taking medication other than antidepressants (
3,
4). Those with chronic depression or current suicidal ideation are also excluded (
1,
5). Consequently, the available “evidence” from clinical trials involves a largely “pure,” uncomplicated population of depressed patients that is rarely seen by most practicing clinicians (
6).
In addition, the care delivered in these efficacy trials, which involves using interviewer-administered measures and frequent and time-intensive follow-up interviews, blinding patients and physicians to treatment, and employing fixed dosing strategies, does not reflect what is and can be done in real-world practices. The available evidence may not translate to the care provided by practicing psychiatrists and primary care physicians (
7). Further, the bulk of the evidence base is for patients who have yet to experience treatment failure in their current episode of depression, even though only about a third of patients achieve remission after a single treatment (
8). Management of most patients after one or more failed treatments is not evidence based.
To address these knowledge deficits, the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study (
www.star-d.org), a large-scale clinical trial funded by the National Institutes of Health, aimed to develop and evaluate feasible treatment strategies to improve clinical outcomes for more representative, “real-world” outpatients with one or more prior failed treatments. The study created its own prospectively defined sample of treatment-resistant patients from a pool of patients currently experiencing a major depressive episode for subsequent inclusion in a series of up to five prospective treatments. Specifically, STAR*D aimed to determine which of several treatments are the most effective “next-step” treatments for patients whose symptoms do not remit or who cannot tolerate the initial treatment and, if needed, ensuing treatments. This article provides an overview of the design, methods, and results of STAR*D, with attention to the implications and limitations of the trial.
The rationale and design of STAR*D
Design
The rationale and design of the study have been fully described elsewhere (
3,
4,
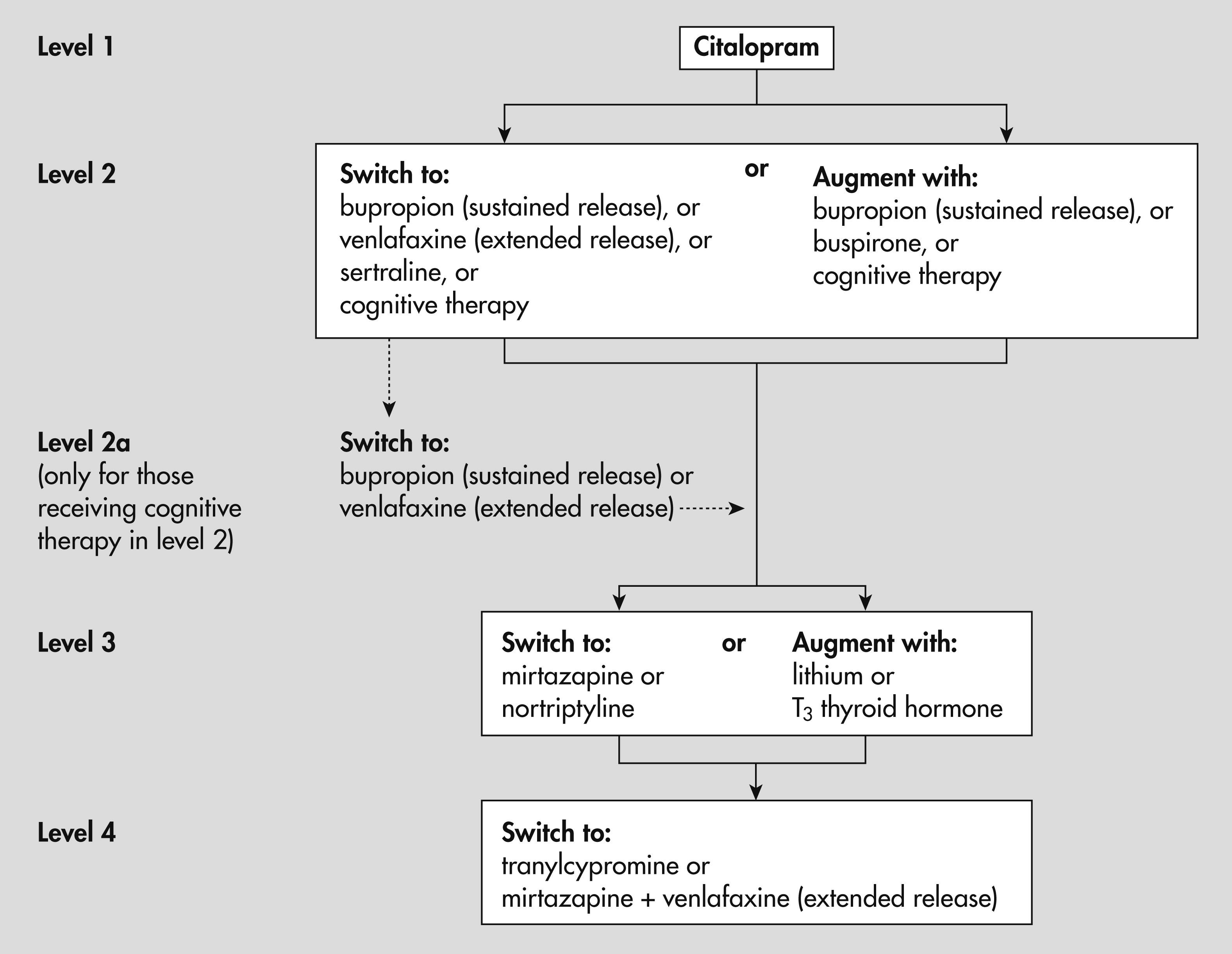
9). STAR*D is the largest prospective clinical trial of major depressive disorder ever conducted. It was a multicenter, nationwide association of 14 university-based regional centers, which oversaw a total of 23 participating psychiatric clinics and 18 primary care clinics. Enrollment began in 2000, with follow-up completed in 2004. All enrolled patients began on a single selective serotonin reuptake inhibitor (SSRI) (citalopram) and were managed by clinic physicians, who followed an algorithm-guided acute phase treatment through five visits over a 12-week course. Dosing was aggressive and focused on maximizing the tolerable dose; if patients who were tolerating a medication had not achieved remission (that is, complete recovery from the depressive episode) by any of the critical decision points (weeks 4, 6, and 9), the algorithm recommended increasing the dose. Patients whose depression did not remit after this initial treatment were able to participate in a sequence of up to three randomized clinical trials or levels. For example, at the end of level 1, patients whose depression had not fully recovered were eligible to participate in level 2 (
Figure 1).
Treatment assignments were made using an equipoise stratified randomized design (
10). To reflect treatment decisions in clinical practice, patients were allowed to choose among acceptable options (for example, to switch to a different treatment or augment the current treatment with an additional treatment). Participants could opt out of certain strategies as long as there were at least two possible options to which they might be randomly assigned.
Participants
Study entry criteria were broadly defined and inclusive. Patients had to have nonpsychotic major depressive disorder identified by clinicians and confirmed with a symptom checklist based on
DSM-IV-TR (
11), for which antidepressant treatment is recommended. Patients, whose ages ranged from 18 to 75, had to score of ≥14 on the 17-item Hamilton Rating Scale for Depression (HAM-D) (
12) and could not have a primary diagnosis of bipolar disorder, obsessive-compulsive disorder, or an eating disorder or have a history of a seizure disorder. A total of 4,041 patients were enrolled in the first level of treatment, making STAR*D the largest prospective clinical trial of depression ever conducted.
Setting
Both primary and specialty care sites that provided care to public- and private-sector patients were selected on the basis of having sufficient numbers of patients, sufficient numbers of clinicians, sufficient administrative support, and sufficient numbers of patients from racial-ethnic minority groups to ensure that the study population would mirror the U.S. census data and that results would be widely generalizable. The median number of clinicians was 14 at the 18 primary care sites and 12 at the 23 specialty sites. Three-quarters of the facilities were privately owned, and approximately two-thirds were freestanding (not hospital based).
Measures
The primary research outcome was the standard definition of remission as measured by the HAM-D (
13). Assessments were conducted by treatment-blinded raters at exit from each treatment level. A secondary instrument, the 16-item Quick Inventory of Depressive Symptomatology—Self-Report (QIDS-SR), was administered at each clinic visit, and remission was measured as a score of ≤5. Because the QIDS-SR was most often successfully collected at a time point closer to when a patient exited a level, the QIDS-SR provided more frequent assessment points during the acute phase and may have been a slightly better reflection of actual remission. The group of patients who improved but whose symptoms did not completely remit was defined as those who showed a ≥50% reduction in QIDS-SR score from baseline to the last assessment in the level.
Intervention
A systematic approach to treatment called measurement-based care was used that can be easily implemented in busy primary care or psychiatric settings (
14,
15). Measurement-based care involves the routine use of symptom and side-effect measurement, with guidance on when and how to modify medication dosages at critical decision points.
STAR*D results
Level 1 outcomes
A total of 2,876 individuals with analyzable data completed level 1 treatment. Measurement-based care was feasible and led to an average citalopram dosage of greater than 40 mg per day, indicating that high-quality care was delivered in these real-world settings. Remission rates were 27% as measured by HAM-D and 33% as measured by QIDS-SR, and response rates were 47% as measured by QIDS-SR. For those whose symptoms remitted, the mean time to remission was approximately 47 days. Factors that increased the chance of remission included being Caucasian, female, and employed and having more years of education and income. Factors associated with lower remission rates were greater chronicity of the current episode, more concurrent psychiatric disorders (especially anxiety disorders or drug abuse), greater degree of general medical comorbidity, and lower levels of functioning and quality of life at baseline.
On average, patients required nearly seven weeks of measurement-based care to achieve remission. Notably, approximately half of the patients who ultimately remitted did so after six weeks, and 40% of those who achieved remission required eight or more weeks to do so (
15).
Level 2 outcomes
After consideration of patient preference, 727 patients were randomly assigned to the switch strategy option in level 2. Nearly one-quarter of patients achieved remission when switched to measurement-based care—guided treatment with sertraline (a “within class” SSRI switch), venlafaxine-XR (a serotonin-norepinephrine reuptake inhibitor), or bupropion-SR (a norepinephrine and dopamine reuptake inhibitor) (
16). Remission rates for bupropion-SR (21% by HAM-D and 26% by QIDS-SR), sertraline (18% and 27%), and venlafaxine-XR (25% for both) were neither statistically nor clinically different by either measure. Mean daily dosage at the final visit for bupropion-SR was 282.7 mg, for sertraline it was 135.5 mg, and for venlafaxine-XR it was 193.6 mg. Of note, the dosage of venlafaxine was less likely to approach the protocol-recommended maximum than that of either of the other two drugs. The overall side effect burden and the rate of serious adverse events did not differ significantly among the three medications.
Moderators of remission were also studied but offered little help in the selection of antidepressants after an initial treatment failure. Neither clinical symptom patterns (including anxious, atypical, and melancholic features) nor standard demographic measures were of clear value in recommending any particular medication for a second step treatment (
17).
Augmentation strategy
After consideration of patient preference, 565 patients were randomly assigned to the augmentation strategy option in level 2. Augmentation of citalopram with bupropion-SR or buspirone led to similar rates of remission as measured by the HAM-D (30% and 30%, respectively) and by the QIDS-SR (39% and 33%, respectively) (
18). However, on an alternative outcome measure, bupropion-SR was associated with a greater total reduction in QIDS-SR scores than buspirone (25% compared with 17%, p<.04). Mean daily dosages at the end of level 2 were 267.5 mg of bupropion-SR and 40.9 mg of buspirone. Of note, augmentation with bupropion-SR was slightly better tolerated than buspirone (intolerable for 13% compared with 21% for buspirone, p<.001). Overall, these results indicate that the choice of either augmentation agent did not produce substantial clinical differences in efficacy.
The data collected did not allow direct comparison of the benefits of switching versus augmenting. Patient preferences were a part of the equipoise randomization strategy, and most patients preferred either augmentation or switching at level 2 (
19). Consequently, patient groups were not equivalent at the point of randomization at the beginning of level 2; the augmentation group at level 2 was somewhat less depressed than the group that switched.
Cognitive therapy
Of those for whom cognitive therapy was acceptable, 182 patients were randomly assigned either to the cognitive therapy switch option or to augmentation of citalopram with cognitive therapy. Remission rates did not differ between those who switched to cognitive therapy (31%) and those who switched medications (31% and 27% remission, respectively) nor were there differences in response or time to remission or response (
20). Switching to cognitive therapy was better tolerated than switching to a different antidepressant. Augmentation results were also similar. Remission rates did not differ between augmentation with cognitive therapy and augmentation with medication (31% and 33% remission). Response rates and tolerability were also similar. However, augmentation of citalopram with medication was more rapidly effective than augmentation with cognitive therapy (40 days compared with 55 days, p<.022).
Level 3
Switch strategy
A total of 235 patients switched medications in level 3. For those whose symptoms did not remit after two antidepressant medication trials, the likelihood of recovery did not differ significantly between patients who switched to mirtazapine and those who switched to nortriptyline (
21). Remission rates for mirtazapine (mean exit dosage of 42.1 mg per day) were 12% as measured by the HAM-D and 8% by the QIDS-SR. The rates for nortriptyline (mean exit dosage of 96.8 mg per day) were 20% and 12%, respectively. QIDS-SR response rates were also similar (13% for mirtazapine and 17% for nortriptyline). Further, tolerability or side-effect burden did not differ significantly between the two treatments.
Consequently, after two consecutive unsuccessful antidepressant trials, a change in pharmacologic mechanism did not affect the likelihood of remission. Also, switching to a third antidepressant single-agent treatment resulted in lower remission rates than in the first two levels.
Augmentation strategy
Medication augmentation was employed for 142 patients in level 3. Similarly, after two failed antidepressant medication treatments (levels 1 and 2), augmentation with a second agent at level 3 was less effective than augmentation at level 2 (
22). Remission rates for lithium augmentation (mean exit dosage of 859.9 mg per day) were 16% as measured by the HAM-D and 13% by the QIDS-SR. For T
3 thyroid hormone augmentation (mean exit dosage of 45.2 micrograms per day) the rates were 25% for both measures. QIDS-SR response rates were 16% for lithium augmentation and 23% for T
3 augmentation. Although these treatment rates did not differ statistically, T
3 was less frequently associated with side effects (p=.045) and with treatment discontinuation because of side effects (23% discontinued compared with 10%, p=.027). When a clinician is considering an augmentation trial, T
3 may have advantages over lithium in effectiveness and tolerability. Further, T
3 offers the advantages of ease of use and no need for blood level monitoring.
Level 4
The switch strategy was employed for 109 patients in level 4. Patients who reached level 4 had failed three aggressive, consecutive, antidepressant trials and had a highly treatment-resistant depressive illness. Remission rates for the combination of mirtazapine (mean dosage of 35.7 mg per day) and venlafaxine-XR (mean dosage of 210.3 mg per day) were 14% as measured by the HAM-D and 16% by the QIDS-SR. For the monoamine oxidase inhibitor tranylcypromine (mean dosage of 36.9 mg per day), rates were 7% by the HAM-D and 14% by the QIDS-SR (
23). Response rates as measured by the QIDS-SR were 24% with the combination and 12% with tranylcypromine. Neither remission nor response rates differed significantly between the combination and tranylcypromine. However, the combination was associated with greater symptomatic improvement and less attrition because of side effects. This comparison is limited by the lower likelihood of an adequate dosage and adequate duration of treatment for patients taking tranylcypromine. Overall, even though clinical outcomes were similar for both groups, the lower likelihood of attrition because of side effect burden and the absence of dietary and concomitant drug restrictions suggest that the combination has some advantages.
Cumulative remission rate and long-term follow-up
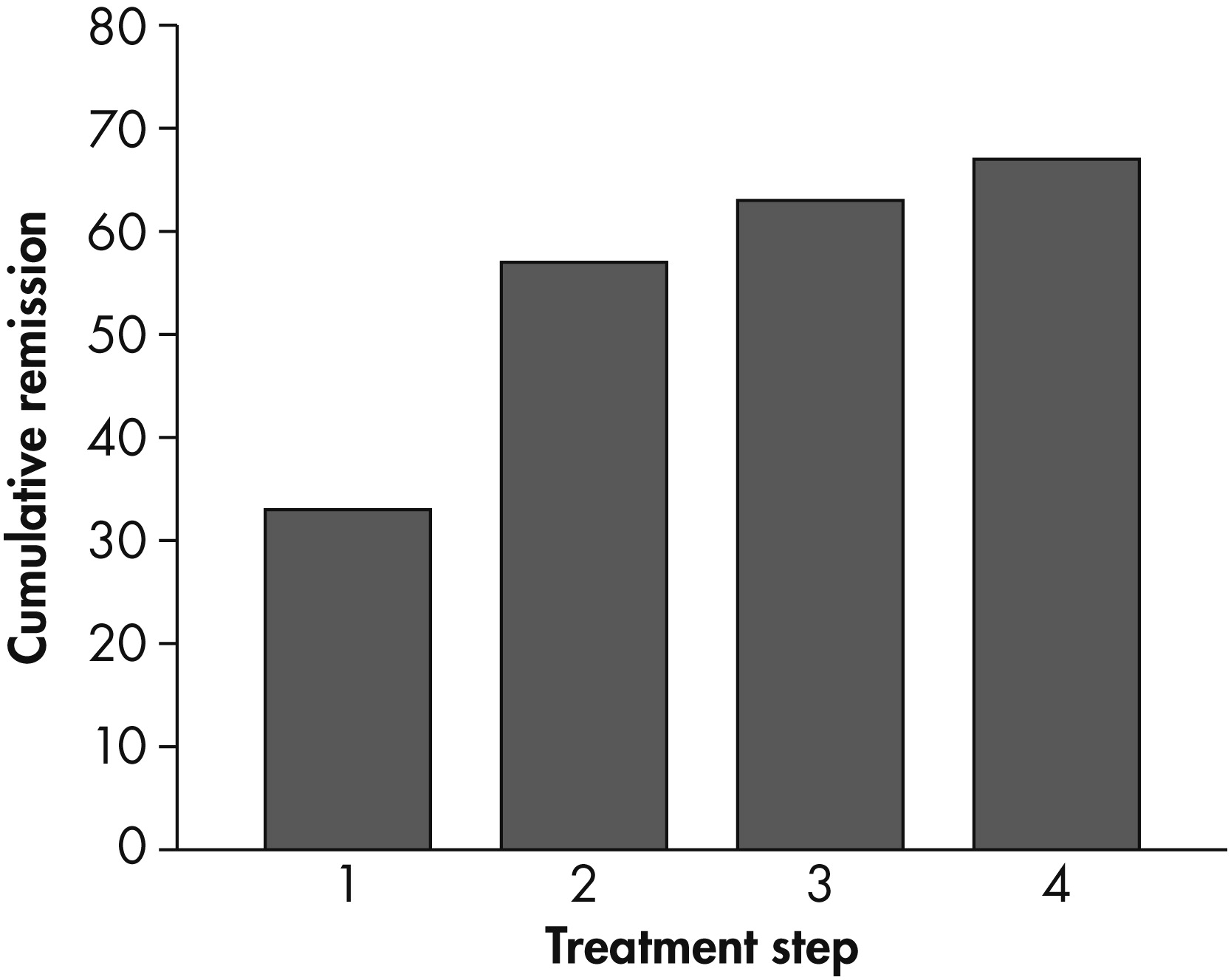
Over the course of the four levels of treatment, the theoretical cumulative remission rate was 67% (see
Figure 2). Remission was more likely to occur during the first two treatment levels (20%–30%) than during levels 3 and 4 (10%–20%).
Patients with a clinically meaningful response, preferably remission, in any of the four levels could enter into a 12-month naturalistic follow-up phase. Those who had required more treatment levels had higher relapse rates during this phase (
24). Also, patients in remission at any level had a better prognosis than those who merely responded, which again provides support for using remission as the preferred aim of treatment.
STAR*D limitations
Although the selection of certain study design elements successfully addressed some primary concerns, such as generalizability and feasibility in real-world practice, the selection came with some clear tradeoffs. First, because patient preference was built into the randomization strategy and patients clearly demonstrated distinct preferences (with the vast majority electing either the switch or augmentation strategies), differences in depressive severity at entrance to the next level and small samples precluded direct comparison of switching and augmenting strategies. Indeed, those who switched to a new medication had more severe illness than those who received augmentation or cognitive therapy.
Thus, if a patient did not achieve remission after treatment in levels 1 and 2, we do not know whether switching medications or augmenting with a second medication led to a better outcome. Similarly, even if a patient had a partial response, STAR*D could not evaluate whether augmentation would have led to a better outcome than switching.
Second, fewer patients than expected selected cognitive therapy, which prevented a more comprehensive assessment of its role. The lower rate of selection of cognitive therapy was likely attributable to the requirement that study participants accept medication (citalopram) as the initial treatment (level 1 entry), which may have biased selection toward individuals who preferred medication. Other likely factors were additional copayments for cognitive therapy or the need to visit an additional provider at another site.
Third, level 1 did not include either a placebo or usual-care control group, which may limit conclusions about remission rates for an initial antidepressant trial. For example, the remission rates approximate what might be expected in eight-week placebo-controlled clinical trials, although such standard efficacy trials do not enroll the diverse population that STAR*D did, which may suggest higher placebo response rates in the traditional trials. However, inclusion of a placebo arm is likely to lead to inclusion of a sample that can limit generalizability of findings, and the aim of STAR*D was not to determine whether treatment is more effective than placebo but rather to show how effective it can be in a representative, community population.
Fourth, the study did not require dosage changes; instead, it used measurement-based care to guide treatment, which reflects use of guidelines in real-world practice. As a result, the trials of STAR*D medications may have been at a lower-than-recommended dosages, as may have happened for some patients who received venlafaxine-XR and tranylcypromine. A difference in the likelihood of having an antidepressant trial at a therapeutic dosage limits the direct comparison of effectiveness of the medications. For example, comparison of venlafaxine at a low-to-moderate dosage and sertraline at a dosage closer to the therapeutic level might unfairly favor a sertraline outcome.
Fifth, the results provide data on the average proportion of patients who are likely to respond to a particular medication or treatment strategy. However, the results do not tell us which patients will respond to which treatments.
Further limitations unrelated to the STAR*D design also can restrict its applicability to current treatments. Since the study was designed approximately a decade ago, not all currently available and employed treatment options were examined. For example, augmentation strategies did not include second-generation antipsychotics, mood stabilizers, or psychostimulants.
Implications of STAR*D findings
STAR*D has key features that define it as an effectiveness trial (
25). Design elements such as broadly inclusive selection criteria and enrollment of patients from primary and specialty settings and with multiple concurrent medical and psychiatric illnesses give STAR*D results high external validity. Comparison of STAR*D participants with the U.S. population highlights the generalizability. The racial-ethnic composition of the enrolled participants approximates that of the U.S. population on the basis of data from the 2000 Census, and the distribution of depressive severity seen in STAR*D participants is consistent with the spectrum reported by Kessler and colleagues (
1) in a nationally representative sample (10% mild, 38% moderate, 39% severe, and 13% very severe). Both facts suggest that the sample was representative of depressed patients in the United States. Further, the participants’ ability to choose which clinic to attend and what treatments were acceptable alternatives mirrors what happens in routine clinical practice, which also enhances the generalizability of these results.
Clinical implications
The primary implications of the STAR*D findings are summarized below.
•.
Remission rates in these representative clinics, in general, were lower than expected on the basis of clinical efficacy trials of antidepressants, which typically report remission rates of 35% to 40% (
9), suggesting the need for several steps to achieve remission for most patients.
•.
There is no clear medication “winner” for patients whose depression does not remit after one or more aggressive medication trials.
•.
Both switching and augmenting are reasonable options for patients after an initial antidepressant treatment has failed.
•.
It may take longer to reach remission than expected, and thus medication trials of at least eight weeks with at least moderately aggressive dosing may be necessary.
•.
Cognitive therapy is a well-tolerated treatment option for patients when an antidepressant treatment fails, and the outcomes patients achieve appear equivalent to those they would have achieved with the trial of a new medication. At the same time, it should be noted that augmentation of citalopram with medication was more rapidly effective than augmentation with cognitive therapy.
•.
Pharmacologic differences between psychotropic medications do not translate into meaningful clinical differences, although tolerability differs.
•.
Neither standard sociodemographic measures nor the symptom patterns that were measured in STAR*D (including anxious, atypical, and melancholic features) predicted a differential benefit from the available switch options at level 2, suggesting that the common practice of selecting treatments based on symptom patterns has little empirical support (
17).
•.
The likelihood of remission after two vigorous medication trials substantially decreases, and remission likely requires more complicated medication regimens for which the existing evidence base is quite thin. Thus an empirically supported definition for treatment-resistant depression seems to be two antidepressant failures.
•.
No statistically significant difference in outcome was found between patients treated in primary care and psychiatric settings when measurement-based care was used in level 1 (
26) or level 2 (
17). Thus primary care physicians, who manage the majority of depressed patients, can be reasonable providers of depression care for at least the first two treatment steps.
•.
The finding that about two-thirds of patients may be expected to reach remission with up to four treatment attempts is encouraging for this disabling illness. Continued treatment attempts, even beyond a second treatment failure, do yield results for some patients.
•.
Longer-term outcomes supported remission as the preferred goal of treatment. During the naturalistic follow-up phase, lower relapse rates were found among participants who entered follow-up in remission than for those who were not (
27).
•.
An important predictor of relapse was greater axis I or III comorbidity. The greater the number of acute treatment steps required from before entry to follow-up (that is, the greater the degree of treatment resistance), the greater the risk of relapse (
27).
Policy implications
STAR*D policy implications are summarized below.
•.
Inclusion of more real-world patients in clinical trials is both feasible and informative. For example, of the group of participants enrolled as a result of the broadly inclusive selection criteria used by STAR*D, only one-fourth would have been enrolled in a standard phase III clinical trial. Results of STAR*’D suggest that broader phase III inclusion criteria would increase generalizability of results to real-world practice, which might reduce placebo response and remission rates (reducing the risk of failed trials) but with some increased risk of adverse events (
6).
•.
The choice of medications for formularies must be carefully considered. Because there was no antidepressant “winner” and the chance of remission did not clearly differ by medication choice, some may argue that formularies can be restricted because of antidepressant equivalence. However, some findings would argue for a broader formulary. For example, antidepressant medications differed in the likelihood of particular side effects, and at this time tolerance cannot be readily predicted. Further, given the multiple treatment steps needed for most participants, availability of a large armamentarium of treatments seems prudent, especially given our inability to predict who will respond to what medication. Finally, given the similar likelihood of response to treatments at level 1 and 2 (some of which have generic formulations) and the inability to predict who will respond better to a particular treatment, available generic antidepressants seem reasonable choices for these first two medication trials.
•.
Measurement-based care—that is, using brief, easy-to-administer instruments to monitor depression severity and side effects, following an evidence-based treatment algorithm, making decisions at key time points, and having remission as a goal of treatment—is a feasible strategy that can be adapted in real-world practice settings—both psychiatric and primary care settings (
14,
15).
•.
Referral guidelines can incorporate the findings that most patients with depressive illness can be adequately treated in primary care for at least two antidepressant trials when measurement-based care is used, thereby reducing the rate of premature referral to psychiatric clinics.
•.
The large number of patients with either recurrent major depressive disorder or with chronic major depressive episodes (>75% in this study), the fact that only about half the patients reached remission after two treatments, and the poor long-term outcomes for patients when two or more acute treatments failed all suggest the need for more evidence to guide the effective treatment of treatment-resistant depression.
Conclusions
STAR*D was a seminal, large-scale, practical clinical trial that provided a great deal of data for clinicians, researchers, and policy makers. The findings are still being actively discussed, analyzed, and disseminated, and the acute-treatment data set is now available in the public domain to allow further analysis. The research infrastructure, which continues as the Depression Trials Network (
www.DTN.com), has completed enrollment for two separate clinical trials whose design was guided, in part, by the findings of STAR*D.
Acknowledgments and disclosures
This project was funded by the National Institute of Mental Health (NIMH) under contract N01MH90003 to the University of Texas Southwestern Medical Center at Dallas. The content of this publication does not necessarily reflect the views or policies of the U.S. Department of Health and Human Services nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. government. NIMH approved the design of the overall study and reviewed its conduct but performed no role in the collection, management, and interpretation of the data analyzed for this report or in the preparation, review, or approval of the manuscript.
Dr. Gaynes has received grants and research support from Bristol-Myers Squibb and Novartis and has served as an advisor for Bristol-Myers Squibb. Dr. Warden has owned stock in Pfizer in the past year. Dr. Trivedi has received consulting fees from AstraZeneca, Bristol-Myers Squibb, Cephalon, Inc., Eli Lilly and Company, Evotec, Fabre-Kramer Pharmaceuticals, Forest Pharmaceuticals, GlaxoSmithKline, Janssen, Johnson and Johnson, Medtronic, Neuronetics, Otsuka Pharmaceuticals, Pfizer, Shire, and Wyeth-Ayerst Laboratories. He has received research support from Targacept. Dr. Wisniewski has received consultant fees from Cyberonics Inc. The other authors report no competing interests.