Genetic Studies
Genetic, molecular, and neuroimaging studies continue to contribute to advances in our understanding of the neurobiological basis of major depressive disorder. However, the extent to which findings from neurobiological studies can help improve the clinical and functional outcome of individuals with the disorder is still uncertain. Thus, in the past 5 years, neurobiological research of depression has become two-tiered to: (1) understand the pathophysiology of the illness; and (2) identify the neurobiological measures for guiding treatment choice.
The identification of single candidate genes associated with major depression has been difficult because of the likelihood that complex psychiatric illnesses are under polygenic influence and are associated with interactions between genetic variants and environmental exposures.
23 One approach has been to go beyond the conventional focus on monoamines in the search for a genetic association.
24 For example, major depressive disorder has been associated with polymorphisms in the glucocorticoid receptor gene
NR3C1,25 the monoamine oxidase A gene,
26 the gene for glycogen synthase kinase-3β (which has a key role in the phosphorylation and regulation of metabolic enzymes and many transcription factors
27), and a group-2 metabotropic glutamate receptor gene (
GRM3).
28 Success has been greater in the identification of candidate genes that are associated with known biological mechanisms and metabolic pathways for antidepressant drugs and, that in turn, can help predict the response from antidepressant treatment.
Many studies have focused on the functional insertion-deletion promoter variant (serotonin transporter-linked polymorphic region [5HTTLPR]) in the serotonin transporter gene (
SLC6A4). A meta-analysis
29 showed two associations, first between the 5HTTLPR long allele and increased response to selective serotonin reuptake inhibitor (SSRI) antidepressant drugs (although not to nortriptyline
30), and reduced side-effects to SSRIs;
31 and second between the 5HTTLPR short allele and increased paroxetine-induced, but decreased mirtazapine-induced adverse effects.
32 Several single-nucleotide polymorphisms (SNPs) in the gene for the serotonin type-2a receptor are associated with outcomes of SSRI treatment.
31,33–35Studies
36–38 of candidate genes associated with other biological mechanisms have emphasised associations between glutamatergic genes (eg,
GRIK4) and citalopram response and adverse effects; between the Met allele of the functional Val/Met polymorphism (rs6265) in brain-derived neurotrophic factor (BDNF) and SSRI response;
39 and between several other BDNF SNPs and desipramine response.
31 Genetic variation in FKBP5—a protein that helps to regulate cortisol binding to the glucocorticoid receptor
40—is associated with antidepressant response;
41,42 whereas genetic variants in
TREK1—a potassium channel mediating SSRI mechanism of action—are associated with non-response to several antidepressants.
43 Genetic variation in the catechol-O-methyltransferase (COMT) gene, which alters COMT activity, is associated with response to treatment with several antidepressants.
44,45Genome-wide association studies further suggest that effectiveness of antidepressants can be predicted by genetic markers other than traditional candidate genes. These genes include those for the corticotrophin-releasing hormone (CRH) receptor-1 (
CRHR1) and CRH binding protein (CRHBP), which predict SSRI response in anxious depression,
46,47 and genes for uronyl-2 sulphotransferase and interleukin-11, which predict response to nortriptyline and escitalopram oxalate, respectively.
48Molecular Studies
At least three main categories of peripheral hormone-type factors, for which genetic variants are associated with major depressive disorder, are implicated in the pathophysiology of the illness: (1) neurotrophic factors and other growth factors, including BDNF, vascular endothelial growth factor, and insulin-like growth factor-1; (2) proinflammatory cytokines, including interleukin-1β, interleukin-6, and tumour necrosis factor-α; and (3) impaired regulation of the hypothalamic-pituitary-adrenocortical (HPA) axis. For example, serum BDNF is decreased in individuals with major depression, and antidepressant treatment reverses this decrease.
49,50The secretion and production of proinflammatory cytokines are increased in individuals who are stressed and depressed,
51,52 and, in major depression, antidepressant drugs can return concentrations of these cytokines to normal or suppress their synthesis.
53 Impaired regulation of the HPA axis in an acute episode of depression has long been documented,
54 and published work has indicated attenuation of neuroendocrine response to the combined dexamethasone-corticotrophin-releasing hormone test—a sensitive measure of altered regulation of the HPA system—during antidepressant treatment.
55Neuroimaging Studies
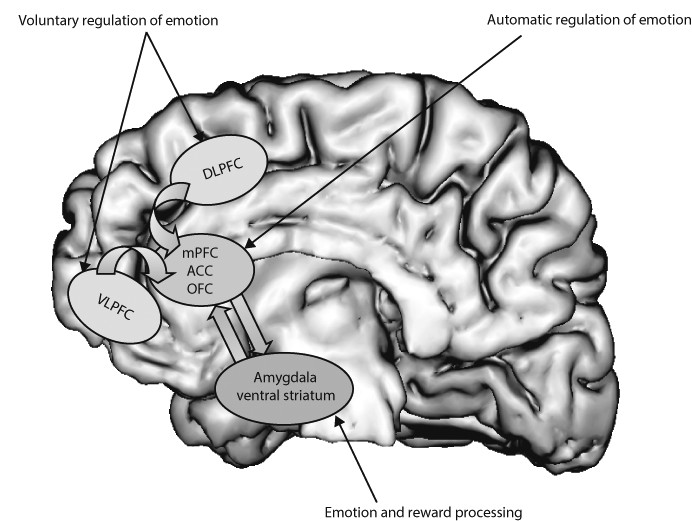
Neural systems that are important to understand major depressive disorder include those that support emotion processing, reward seeking, and regulate emotion, all of which are dysfunctional in the disorder. These systems include subcortical systems involved in emotion and reward processing (eg, amygdala, ventral striatum); medial prefrontal and anterior cingulate cortical regions involved in processing emotion and automatic or implicit regulation of emotion; and lateral prefrontal cortical systems (eg, ventrolateral prefrontal cortex and dorso-lateral prefrontal cortex) involved in cognitive control and voluntary or effortful regulation of emotion.
56 These systems can be conceptualised as a medial prefrontal-limbic network, including amygdala, anterior cingulate cortex, and medial prefrontal cortex that is modulated by serotonin neurotransmission,
57–59 and a reward network centred on ventral striatum and interconnected orbito-frontal and medial prefrontal cortices that is modulated by dopamine (
figure 1).
60Neuroimaging studies of major depressive disorder
61–65 have provided evidence for specific functional abnormalities in these neural systems in adults. Converging findings from these studies suggest abnormally increased amygdala, ventral striatal, and medial prefrontal cortex activity, mostly to negative emotional stimuli, such as fearful faces. Abnormally reduced ventral striatal activity to positive emotional stimuli,
61,66 and during receipt and anticipation of reward in adults
67 and adolescents
68 with depression have also been reported. These findings support a bias of attention towards negative emotional stimuli, and away from positive emotional and reward-related stimuli in individuals with major depression. Further studies are needed to examine neural activity in the prefrontal cortical regions that are implicated in voluntary and implicit regulation of emotion. These functional neuroimaging studies are complemented by increased findings suggesting reductions in grey matter in key regions of these neural systems (including amygdala
69), age-related decreases in volume of the anterior cingulate cortex,
70 and post-mortem findings indicating neural-cell and glial-cell pathology in prefrontal cortical regions.
71Additional evidence for abnormalities in neural systems for emotion and reward processing and regulation of emotion in major depressive disorder has come from neuroimaging studies of emotion processing, which used measures of functional connectivity, including effective connectivity—ie, the effect that activity in one region exerts over that in another. For example, one study
72 reported abnormal-inverse effective connectivity in medial prefrontal cortical-amygdala during emotion labelling of happy faces in individuals with major depression. These findings suggest abnormally increased top-down regulation of the amygdala by medial prefrontal cortex in these individuals, which again suggests a bias away from attendance to positive emotional stimuli. Other studies
73 have applied diffusion tensor imaging, which uses measures of the diffusivity of water in different directions—either parallel or perpendicular to the direction of white-matter fibres—to construct whole-brain measures of patterns of white-matter connectivity between different neural regions. Findings from these studies, have shown abnormal prefrontal cortico-subcortical white-matter connectivity between regions supporting emotion regulation in adults with depression.
73Examination of brain activity during rest can provide valuable insights into how the brain functions during self-reflection, which could be abnormal in individuals with major depression. Studies have provided evidence for a malfunction of the default mode network.
74 This network includes several midline regions, including the inferior (ventral) part of the medial prefrontal cortex (vmPFC). Anterior parts of this network (eg, vmPFC) are involved in self-referential processing
74 and tend to deactivate during cognitive tasks as cognitive resources are redirected. The normal decrease in vmPFC activity during difficult tasks might indicate intact emotional gating and, conversely, absence of this deactivation might indicate impaired gating. Findings from analysis of task-independent deactivations suggest an abnormality of the default mode network in vmPFC in major depressive disorder. A method to examine resting-state-related neural activity is measurement of the correlation of low-frequency blood oxygen level-dependent temporal fluctuations (LFBF) in steady-state functional MRI data between different neural regions. One study
75 showed that individuals with depression had increased resting-state connectivity in medial prefrontal cortex regions in the default mode network, including the anterior cingulate cortex and the vmPFC.
A meta-analysis
76 of several neuroimaging studies of major depressive disorder identified two neural systems of importance to the illness. One network that centred on the dorsolateral prefrontal cortex and more dorsal regions of the anterior cingulate cortex, but that also included other regions implicated in cognitive control, was characterised by reduced activity in the resting state, which returned to normal with treatment. A second network, centred on medial prefrontal cortex and subcortical regions, was hyperactive to emotional stimuli in the depressed state, but returned to normal after antidepressant treatment. This meta-analysis
76 provides further evidence of increased activity in neural systems supporting emotion processing (amygdala and medial prefrontal cortex), and reduced activity in neural systems supporting regulation of emotion (eg, dorsolateral prefrontal cortex).
Neuroimaging studies have also examined neural predictors of outcome and neuroimaging measures that change with response to different antidepressant treatments. These studies have focused mainly on the role of the serotonergically-mediated medial prefrontal-limbic network. In major depression, SSRI drugs modulate emotion-induced activity in this network, and response to SSRIs is predicted by activity in this region.
77 LFBF correlations between subcortical regions and the anterior cingulate cortex can also increase after SSRI treatment in individuals with depression.
78,79 However, less is known about the effects of non-serotonergic drugs on the medial prefrontal-limbic network, and the extent to which pretreatment activity in this region predicts subsequent response to non-serotonergic antidepressants needs to be examined. Desynchronisation of the anterior cingulate cortex, and its functional connectivity with the amygdala predicts rapid antidepressant response to ketamine,
80 but response to cognitive behavioural therapy is predicted by reduced medial prefrontal activity, and increased amygdala activation.
81 Response to deep-brain stimulation correlates with reduced activity in ventral (subgenual) regions of the anterior cingulate cortex,
82,83 and increased metabolism in ventral striatum.
83Integration of Measures
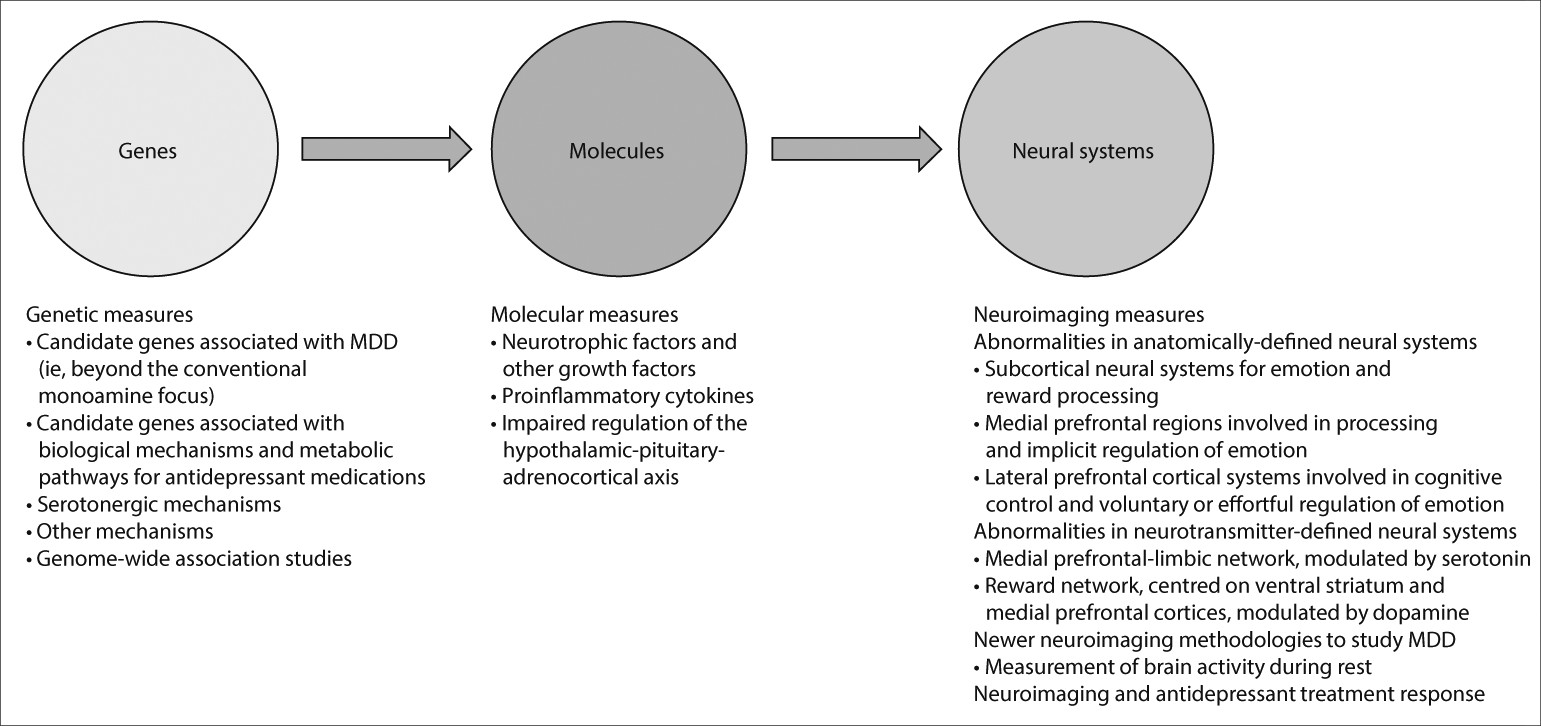
Studies examining a combination of genetic, molecular, and neuroimaging measures to identify relations among genes, molecules, neural systems, and behaviour in major depressive disorder could increase our understanding of the underlying pathophysiological processes and prediction of treatment response (
figure 2). Relations between genetic variation in neutrophic factors, and variation in concentrations of these factors, such as BDNF and vascular endothelial growth factor, and structural and functional variation in neural regions supporting mood regulation and associated behaviours and neurocognitive function, are well documented.
84–88 This variation might in turn reduce the ability to respond to treatments dependent on intact neurocognition, such as cognitive behavioural therapy.
Reports
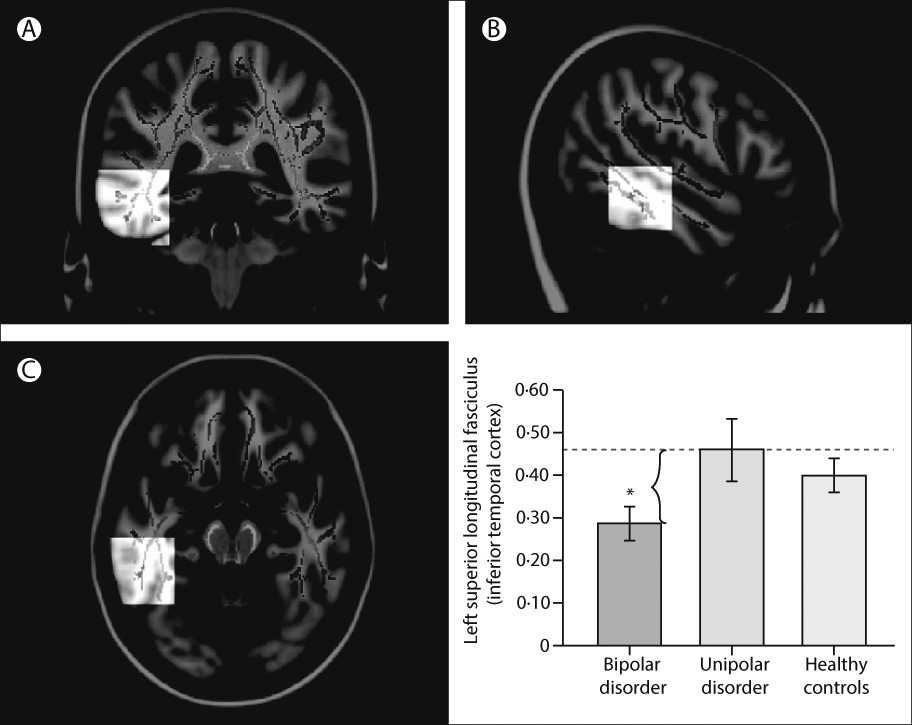
89–93 have emphasised associations between genetic variation in function of the HPA axis, genetic variation between—and variation in concentrations of—inflammatory cytokines, and functional and structural variation in key neural regions supporting mood regulation. Studies have also indicated how neuroimaging techniques can be applied to diagnosis and how specific neuroimaging measures might help to distinguish major depressive disorder from bipolar depression.
72,94,95 In turn, these measures can facilitate early diagnosis and can inform treatment choice (
figure 3). Computerised machine learning and techniques for pattern recognition in combination with neuroimaging techniques can be used to classify individuals, case-by-case, into diagnostic groups on the basis of neuroimaging measures.
64,96