A Clinical Guide to Frontotemporal Dementias
Abstract
The term frontotemporal dementia (FTD) describes a diverse group of clinical syndromes, including behavioral-variant FTD (bvFTD), nonfluent/agrammatic-variant primary progressive aphasia (nfvPPA), semantic-variant primary progressive aphasia (svPPA), FTD motor neuron disease (FTD-MND), progressive supranuclear palsy syndrome (PSP-S), and corticobasal syndrome (CBS). Although each of these syndromes may be distinguished by their respective disturbances in behavior, language, or motor function and characteristic imaging findings, they may present a diagnostic dilemma when encountered clinically. In this article, we review the clinical features, diagnostic criteria, pathology, genetics, and therapeutic interventions for FTD spectrum disorders.
Frontotemporal lobar degeneration (FTLD) is a pathologically defined entity involving synapse loss, gliosis, neuronal loss, and ultimately gross atrophy within the frontal and anterior temporal lobes, basal ganglia, and thalamus (1). FTLD gives rise to a heterogeneous group of clinical syndromes collectively referred to as frontotemporal dementia (FTD). FTD may be subdivided into three distinct core clinical syndromes, including behavioral-variant FTD (bvFTD) and two varieties of primary progressive aphasia (PPA), the nonfluent/agrammatic-variant primary progressive aphasia (nfvPPA) and the semantic-variant primary progressive aphasia (svPPA). Patients with svPPA may be further divided into left and right temporal variants. Patients with chiefly right hemispheric atrophy (including those with bvFTD and the right temporal svPPA) tend to present with predominantly behavioral syndromes, whereas patients with chiefly left hemispheric atrophy (including those with nfvPPA and left temporal svPPA) tend to present with a language-dominant syndrome. The core clinical variants of FTD overlap with a wider set of clinical FTD spectrum disorders, including three additional syndromes with prominent motor features: FTD motor neuron disease (FTD-MND), progressive supranuclear palsy syndrome (PSP-S), and corticobasal syndrome (CBS).
FTD is the most common form of dementia occurring in patients under age 65. Several cohort studies have established the incidence and prevalence of FTD to be on par with Alzheimer’s disease (AD) among patients from ages 45 to 65 (2–4). Several studies on FTD report an incidence of around four per 100,000 person-years (2, 5). The prevalence of FTD is reported to be between 10 and 20 per 100,000, although estimates have ranged widely (3, 6, 7). It should be noted that the prevalence of FTD is greatly underestimated, given decreased recognition among providers who are more familiar with better recognized psychiatric or neurological diseases. The mean age of onset for FTD is typically reported to be late in the sixth decade of life, and the mean ages of onset for PSP-S and CBS are in the seventh decade (4, 6, 7). Although a male predominance has been reported in FTD (3), other large cohort studies have established a roughly equal incidence between females and males (4, 8–10).
FTD has a high incidence of heritability compared with many other neurodegenerative dementias. About 40% of patients with FTD have a family history of dementia within a first-degree relative, with approximately 15% showing a family history suggestive of an autosomal-dominant mutation associated with their disease (6, 8, 11, 12). Heritability is not, however, uniform across the clinical syndromes. Patients with FTD-MND, for instance, have the strongest family histories of neurodegenerative disease, whereas patients with svPPA tend to have the least suggestive family histories (11). Survival is also variable among the FTD syndromes. The mean survival after onset of disease is about 8.7 years for patients with FTD and 9.4 years among patients with PPA, but only around 3 years for patients with FTD-MND (6, 13). Unfortunately, the mean duration from onset to diagnosis of FTD syndromes is 2.5 years, so patients may be well along their clinical course by the time a formal diagnosis is made (6).
Classification schemas for FTD do not perfectly reflect the rich diversity of clinical phenotypes and underlying pathologies associated with FTLD. Even among patients with the same clinical diagnosis, symptoms and underlying pathologies are heterogeneous. In addition, there is increasing syndromic overlap among all patients with FTD as the disorder progresses (14). Discrete clinical diagnoses may be made with attention to early symptomatology. Some authors suggest inclusion of secondary diagnoses as patients accumulate features that meet criteria for alternate syndromes (15). A patient with early language changes followed by features of CBS, for instance, may be given a designation of PPA-CBS in the course of his or her disease. A patient’s initial clinical syndrome and subsequent progression of clinical features may inform clinicians of potential underlying neuropathology. The earliest stages of progression in FTD involve dysfunction in neuronal networks involved in functions, such as self-monitoring, evaluation of internal mental states, awareness of emotion, sustaining personality, and production of language (16). These disrupted neural networks involve central hubs of function correlating with the earliest sites of pathology and neurodegeneration in FTLD (17). The typical sites of early pathology are distinct among particular pathologic categories of FTLD, and these FTLD categories may be distinguished by the presence of neuronal or glial accumulations of specific misfolded proteins on autopsy.
It is unclear if these hallmark proteins are a driving force or simply a repercussion of a process that confers selective vulnerability within specific cell types and specific networks in FTLD. Various FTLD-associated proteins are, however, hypothesized to be a driving force of pathologic progression by means of a prion-like spread among cells in a network-dependent fashion (18). Ideally, a clinician may use observation of clinical symptoms, associated patterns of atrophy on imaging, and potentially other available biomarkers to shed light on the particular anatomy of involvement in a patient with FTD and by this method determine the likelihood of a specific underlying pathology.
Behavioral-Variant Frontotemporal Dementia
Clinical Presentation of bvFTD
bvFTD involves a progressive disturbance in personality, emotion, and behavior (19). The diverse array of aberrant behaviors in bvFTD (including apathy, disinhibition, changes in empathy, compulsions, and dietary changes) reflects a decline in function and network connectivity among several paralimbic brain regions, including the medial frontal, orbitofrontal, anterior cingulate, and frontal insular cortices (20, 21). Although many neurodegenerative and psychiatric diseases have behavioral features, in bvFTD the behavioral features are the earliest, most salient, and most disabling aspect of the clinical syndrome. The clinical symptoms of bvFTD reflect diminished function in the medial frontal, insular, and anterior temporal regions, with symptoms driven by the severity of the dysfunction in the nondominant frontotemporal regions.
Apathy is the most common first symptom among patients with bvFTD (19, 22). Symptoms may manifest early with new passivity, indecisiveness, and decreased libido. Patients later experience a more profound loss of interest in hygiene tasks, hobbies, social interactions, work responsibilities, and domestic responsibilities, leading to marked disability and loss of independence. Inertia may be observed as an overall paucity in spontaneous action, and patients may require constant prompting to groom, bathe, converse, or engage in physical activity. Loss of motivation and initiative in bvFTD best correlates with network dysfunction and atrophy involving the dorsomedial frontal regions, including the anterior cingulate and adjacent prefrontal cortices (20, 22–24).
Early behavioral disinhibition is the second most common feature of bvFTD, although it can give rise to the most obvious, upsetting, and disruptive features of the disease. Patients experience a decrement in their ability to weigh immediate rewards against punishments and long-term goals, correlating to atrophy and disrupted connectivity in orbitofrontal and subgenual cingulate cortices, chiefly on the right (20, 25, 26). Dysfunction within the pregenual portion of the anterior cingulate also contributes to a loss of constraint from embarrassment and self-consciousness (27). In addition, disinhibition also may occur in regard to a loss of disgust and aversion to typically repellant stimuli (such as old discarded food, garbage, and bodily fluids), correlating to anterior insular cortex dysfunction (28, 29). Social disinhibition can result in inappropriate cordiality and physical affection with strangers, inappropriate touching, and unwanted sexual advances. Hypersexual behavior is relatively rare in bvFTD; the overwhelming majority of patients experience a decrease in affection and are often profoundly hyposexual for years before the diagnosis of their illness (30).
Criminal behaviors are unfortunately common and may include speeding, theft, public urination, public nudity, and assault. Patients experience loss of social decorum and may become profane, unkempt, and malodorous. They may lack the restraint to wait in line, wait for their turn to speak in conversation, or respect the personal space of others. Inappropriate mirth and jocularity are occasionally observed, often involving childish simple humor and compulsive use of puns (31). Disinhibition also manifests as increased impulsivity, resulting in reckless driving, excessive spending, and overly forthcoming comments (such as blurting out Social Security numbers and bank passwords to strangers). Typically, bvFTD patients experience a profound, obvious loss of insight regarding their behavioral changes (32). They are generally unaware or ambivalent toward the appropriateness of their actions and the subjective experience of others to them. They are easily manipulated and are frequently swindled out of their money.
Social function of patients with bvFTD may be further impaired by a loss of empathy and sympathy. Family members describe loss of personal warmth, decrease in affection, decrease in social interest, ambivalence toward the emotions of others, and profound selfishness, even in the wake of family tragedy. A decrease in empathy has been correlated to atrophy within the right frontoinsular cortex, right subgenual cingulate, right anterior temporal lobe, and right ventral striatum (33, 34). Patients with right anterior temporal atrophy (as in the case of right temporal svPPA) in particular display an inability to interpret the emotions of others around them. Interpersonally, these patients may appear cold and unresponsive. Other patients may smile often and project a childish demeanor, although their expressiveness is disconnected from the context of emotions of others. This can be particularly evident at events like funerals or religious ceremonies where behavioral restraint is expected.
Compulsive behaviors occur in a variety of forms among patients with bvFTD (19). Simple, stereotyped, repetitive behaviors may include tapping, picking, rocking, humming, throat clearing, and other perseverative motor tasks. Patients also present with stereotypies of speech, including repetition of words, phrases, and stories. More complex compulsive behaviors may include hoarding, repetitive checking, and rituals involving cleaning, walking repetitive routes, taking excessive trips to the bathroom, and arranging items. Care should be taken to distinguish ritualized walking habits from the pacing and akathisia that may occur from psychotropic medications. Voxel-based morphometry studies that capture regional atrophy show correlations of obsessive behaviors in bvFTD to a variety of cortical and striatal regions, including the bilateral globus pallidus, left putamen, and lateral temporal lobes (35). Aberrant motor behavior in particular has been correlated with atrophy of the supplementary motor cortex in the right hemisphere (20).
Hyperorality and dietary changes represent another core feature of bvFTD. Patients may experience compulsive eating, including binge eating regardless of satiety (36). Alteration of food preferences is common, most often leading to a preference for carbohydrates (37). Compulsive consumption habits are not limited to food, as some patients exhibit new excessive alcohol or tobacco use. Others eat inedible objects, such as buttons. The onset of hyperorality best correlates to disrupted connectivity and degeneration within the right orbitofrontal cortex, insular cortex, ventral striate, and ventral hypothalamus (36, 37). As mentioned earlier, dysfunction within the insular cortex is associated with a loss of disgust and may be responsible for consumption of spoiled food and garbage. Additionally, temporal lobe involvement may give rise to narrow or rigid and sometimes ritualistic food choices (38).
Formal Neuropsychological Testing in bvFTD
Patients with bvFTD have relative deficits in tests of executive function that tap abilities in working memory, cognitive control, attention, flexibility, generation, and abstraction (19, 39). Formal neuropsychological testing is, however, often unrevealing of early stages of bvFTD, as paralimbic pathology typically precedes dysfunction within the dorsolateral prefrontal cortex, a region involved in standard executive tasks, such as sorting and set shifting (40). Testing of social function, including tests involving faux pas awareness, interpretation of emotion, embarrassment, and theory of mind, may be sensitive in distinguishing patients with bvFTD from healthy individuals, although these tests are rarely implemented in clinical practice and not included in current diagnostic criteria (19, 27, 41). Performance in additional cognitive domains, such as visuospatial function and memory, may be relatively intact compared with executive and social function, although these additional domains are not necessarily spared. Although memory consolidation deficits in bvFTD may not be as profoundly impaired as they are in AD, patients with bvFTD experience measurable memory decline, particularly in late stages of disease (39, 42). Moreover, functional clinical measures (reported by family member assessment) show no distinction between FTD and AD in terms of reported disability in memory (43).
Imaging Findings in bvFTD
Brain imaging may support a clinical diagnosis of bvFTD, although imaging may be normal in the earliest stages of disease (44). MRI imaging in cases of bvFTD typically reveals disproportionate frontal, insular, or anterior temporal lobe atrophy, particularly on the right (19, 21, 44, 45). Given the early network dysfunction associated with bvFTD, fMRI may establish selective anterior cingulate and frontoinsular network dysfunction, although this tool is not available clinically (46). Fludeoxyglucose (18F) positron emission tomography (FDG-PET) and less frequently used single-photon emission computed tomography (SPECT) are clinically available and may reflect frontotemporal hypometabolism distinguishing bvFTD from other neurodegenerative diseases (47, 48). Changes on FDG-PET may precede changes on MRI. In fact, the distinction between AD and FTD remains the sole indication for the use of central nervous system FDG-PET in dementia to be reimbursed by the Centers for Medicare and Medicaid Services. FDG-PET and SPECT are rarely necessary, however, because they seldom add additional resolution to findings already apparent on MRI.
Diagnosis of bvFTD and Distinction From Other Syndromes
A clinical diagnosis of possible bvFTD requires progressive deterioration of behavior or cognition and must involve at least three core clinical features as detailed earlier, including early behavioral disinhibition, early apathy-inertia, early loss of sympathy-empathy, obsessive behaviors (including early perseverative, stereotyped, or compulsive-ritualistic behavior), prominent dietary change, and suggestive neuropsychiatric testing (with relative sparing of episodic memory or visuospatial skills; Box 1) (19). A designation of probable bvFTD requires satisfaction of clinical criteria for possible bvFTD, along with a caregiver report of functional decline and suggestive imaging findings, including frontal or anterior temporal atrophy on MRI or CT scan (Figure 1) or frontal or anterior temporal hypoperfusion or hypometabolism on PET or SPECT. Although these diagnostic criteria are relatively sensitive and specific to bvFTD, there are some pitfalls and clinical nuances that may make diagnostic clarity challenging. One-fifth of patients with bvFTD will have parkinsonism on examination, and 12.5% will meet criteria for CBS or PSP-S during their clinical course (12, 19). These diagnoses are not mutually exclusive, in that they represent alternate or coexisting phenotypes of the same potential underlying pathologies.
Box 1: INTERNATIONAL CONSENSUS CRITERIA FOR BEHAVIORAL-VARIANT FRONTOTEMPORAL DEMENTIAa
I. Neurodegenerative disease
The following symptom must be present to meet criteria for behavioral-variant frontotemporal demential (bvFTD):
A.
Shows progressive deterioration of behavior and/or cognition by observation or history (as provided by a knowledgeable informant).
II. Possible bvFTD
Three of the following behavioral/cognitive symptoms (A–F) must be present to meet criteria. Ascertainment requires that symptoms be persistent or recurrent, rather than single or rare events.
A.
Early behavioral disinhibition [one of the following symptoms (A.1–A.3) must be present]:
A.1.
Socially inappropriate behavior
A.2.
Loss of manners or decorum
A.3.
Impulsive, rash or careless actions
B.
Early apathy or inertia [one of the following symptoms (B.1–B.2) must be present]:
B.1.
Apathy
B.2.
Inertia
C.
Early loss of sympathy or empathy [one of the following symptoms (C.1–C.2) must be present]:
C.1.
Diminished response to other people’s needs and feelings
C.2.
Diminished social interest, interrelatedness or personal warmth
D.
Early perseverative, stereotyped, or compulsive/ritualistic behavior [one of the following symptoms (D.1–D.3) must be present]:
D.1.
Simple repetitive movements
D.2.
Complex, compulsive or ritualistic behaviors
D.3.
Stereotypy of speech
E.
Hyperorality and dietary changes [one of the following symptoms (E.1–E.3) must be present]:
E.1.
Altered food preferences
E.2.
Binge eating, increased consumption of alcohol or cigarettes
E.3.
Oral exploration or consumption of inedible objects
F.
Neuropsychological profile: executive/generation deficits with relative sparing of memory and visuospatial functions [all of the following symptoms (F.1–F.3) must be present]:
F.1.
Deficits in executive tasks
F.2.
Relative sparing of episodic memory
F.3.
Relative sparing of visuospatial skills
III. Probable bvFTD
All of the following symptoms (A–C) must be present to meet criteria.
A.
Meets criteria for possible bvFTD
B.
Exhibits significant functional decline (by caregiver report or as evidenced by Clinical Dementia Rating Scale or Functional Activities Questionnaire scores)
C.
Imaging results consistent with bvFTD [one of the following (C.1–C.2) must be present]:
C.1.
Frontal and/or anterior temporal atrophy on MRI or CT
C.2.
Frontal and/or anterior temporal hypoperfusion or hypometabolism on PET or SPECT
IV. bvFTD with definite frontotemporal lobar degeneration (FTLD) pathology
Criterion A and either criterion B or C must be present to meet criteria.
A.
Meets criteria for possible or probable bvFTD
B.
Histopathological evidence of FTLD on biopsy or at postmortem
C.
Presence of a known pathogenic mutation
V. Exclusionary criteria for bvFTD
Criteria A and B must be answered negatively for any bvFTD diagnosis. Criterion C can be positive for possible bvFTD but must be negative for probable bvFTD.
A.
Pattern of deficits is better accounted for by other nondegenerative nervous system or medical disorders
B.
Behavioral disturbance is better accounted for by a psychiatric diagnosis
C.
Biomarkers strongly indicative of Alzheimer’s disease or other neurodegenerative process
___________________
aAdapted from Rascovsky et al. (19)
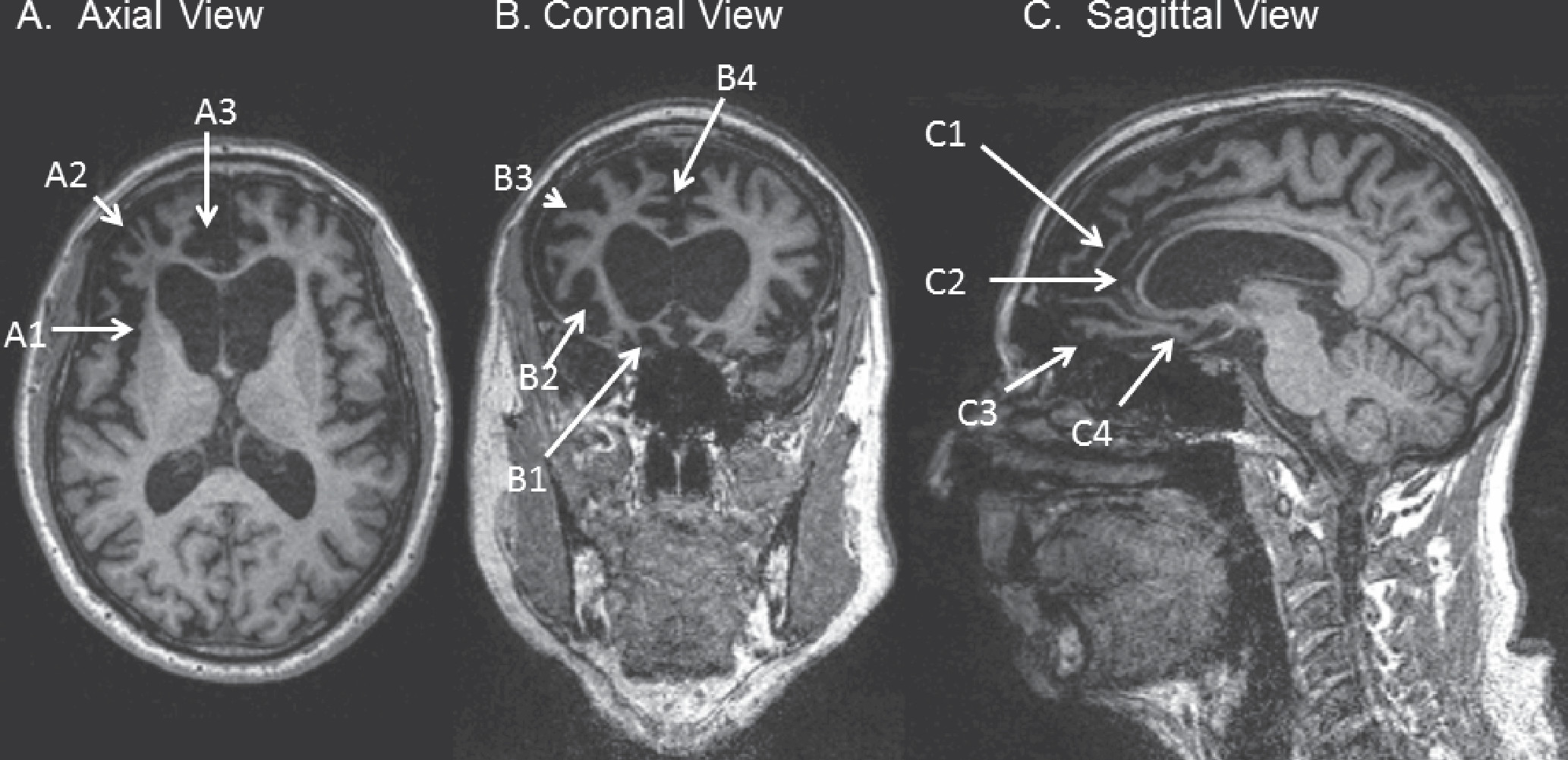
Figure 1. T1-Weighted MRI of a 62-Year-Old Patient With Behavioral-Variant Frontotemporal Dementia (bvFTD)
A: Axial view at the level of the genu of the corpus callosum in radiological orientation, showing asymmetric, right greater than left, atrophy of the frontal lobe, including the insular cortex (A1), lateral prefrontal cortex (A2), and anterior cingulate along with adjacent medial prefrontal cortex (A3). B: Coronal view at the level of the rostral corpus callosum in radiological orientation, showing asymmetric atrophy of the orbitofrontal cortex (B1), insular cortex (B2), lateral prefrontal cortex (B3), and anterior cingulate along with adjacent medial prefrontal cortex (B4). C: Sagittal view of the mesial right hemisphere, showing frontal atrophy, including mesial prefrontal cortex (C1), pregenual cingulate (C2), orbitofrontal cortex (C3), and subgenual cingulate (C4).
The presence of underlying AD pathology is by convention an exclusionary criterion for a diagnosis of bvFTD. AD pathology may, however, give rise to a frontal AD syndrome that meets full clinical criteria for bvFTD (49). Fortunately, several clinical and radiographic tools may aid in the distinction between bvFTD and patients with AD presenting with a bvFTD phenotype. The frontal-variant AD cohort will, for instance, have a greater burden of parietal and mesial temporal atrophy compared with patients with bvFTD. Patients with frontal AD also tend to have a greater degree of memory disturbance and executive dysfunction early in their disease course but less severe behavioral disturbances (49). FDG-PET may aid a clinical diagnosis by distinguishing the frontotemporal pattern of hypometabolism seen in FTD from the temporoparietal and posterior cingulate hypometabolism seen in AD (48). Amyloid biomarkers provide the most definitive diagnostic clarity to distinguish between FTD and AD in patients. Cerebrospinal fluid (CSF) studies are commercially available and can distinguish patients with AD pathology by confirming decreased amyloid beta and increased tau in CSF (50, 51). Amyloid PET, using florbetapir (18F) or Pittsburgh compound B (PiB) ligands, may also be used to confirm high amyloid beta deposition in cortical regions, consistent with a diagnosis of AD pathology rather than FTLD (51). Unfortunately, although amyloid PET is less invasive than a lumbar puncture and is an FDA-approved procedure, it is rarely covered by insurance. Interpretation of amyloid-based modalities may be challenging for patients over 70, given that the false positive rate increases with age (52).
Dementia with Lewy bodies (DLB) occasionally presents a diagnostic challenge among patients with behavioral syndromes. Patients with DLB often experience challenging behaviors and personality changes related to delusions and depression. Patients with DLB rarely meet the bvFTD diagnostic criteria outlined above. Patients with DLB may also be distinguished by their hallmark features of fluctuating attention and visual hallucinations, because psychosis is exceedingly rare in sporadic bvFTD (53). Additional hints of Lewy body disease include the presence of a preclinical prodrome, such as anosmia, REM behavior disorder, depression, and dysautomonia (including erectile dysfunction, syncope, and constipation) (54).
bvFTD patients show considerable overlap with psychiatric disorders. The term bvFTD phenocopy is typically applied to patients with the core clinical features of bvFTD in the absence of clinical progression or imaging changes (55). Previous speculation about this patient group has included discussion of autism spectrum disorders and late-life psychiatric disease. More recent studies have, however, linked cases of bvFTD phenocopy to known autosomal-dominant causes of FTD, such as pathologic repeat expansion of the C9ORF72 gene (56). Other examples of confusion between psychiatric illness and FTD can be found within the early symptomatology and clinical prodrome of patients with bvFTD. Apathy often occurs alone in the earliest stages of bvFTD and can easily be mistaken for depression. In a literature review of bvFTD cases published between 1990 and 2007, amounting to 751 individuals, 6% of patients presented with psychosis early in their disease course, and over 60% of these cases met criteria for formal diagnoses of bipolar disorder, schizophrenia, schizoaffective disorder, or depression with psychosis (57). The mean age of onset of this subgroup was 40.1 years. Carriers of autosomal-dominant FTD risk factor genes are also known to present with prodromal psychiatric features. Patients with pathologic repeat expansions within C9ORF72 frequently experience prodromal psychotic symptoms, including somatoform delusions such as delusions of infestation (58, 59). Variability within the GRN gene is also associated with occasional prodromal psychosis in FTD and risk of psychiatric illness, including schizophrenia and bipolar disease (59–61).
Primary Progressive Aphasias
The term primary progressive aphasia (PPA) refers to clinical syndromes in patients with progressive neurodegeneration resulting in a disturbance in language production and/or comprehension. In PPA, language must be the earliest feature of disease and the principal cause of disability (62). PPA is divided into three distinct clinical variants, each with its own unique deficits, anatomical features, and pathological correlates. Two of the three forms of PPA—svPPA and nfvPPA—fit within the umbrella of FTD. The third form of PPA, logopenic-variant PPA (lvPPA), is typically a language-predominant presentation of AD and therefore is not discussed in this article (63).
Semantic-Variant Primary Progressive Aphasia (svPPA)
Patients with svPPA experience disrupted network connectivity involving the anterior temporal lobes (64). The anterior temporal lobes are central hubs that allow retrieval of the semantic memory (a person’s library of information about objects, animals, and people around them). Current clinical criteria pertain to the more common left temporal variant of svPPA, which results in more obvious changes in language and concrete knowledge. Early presentations of the right temporal variant of svPPA are typically associated with a unique behavioral syndrome (which may meet formal criteria for bvFTD); patients exhibit deficits in knowledge about familiar people and display severe problems with empathy. Although temporal lobe asymmetry drives the differing symptom profiles of the svPPA variants, both variants increasingly overlap as patients develop increasing contralateral temporal lobe pathology late in disease progression (65). Patients with svPPA have some of the longest survivals among FTD patients, with a mean survival of 9.1 years postdiagnosis (6). Left-handedness is overrepresented among patients with svPPA, suggesting that nonnormal lateralization may be associated with some intrinsic risk for developing the syndrome (66). Presentations of svPPA are almost never familial (67).
Progressive anomia, a deficit in naming, is the principal feature at onset of the left temporal variant of svPPA. Family members may report this change as increasingly vague and circumlocutory speech, with increasingly few specific nouns (39). Deficits start in low-frequency words and progress to words of higher frequency. Furthermore, patients with svPPA are less likely to provide conceptual information about a missed word compared with other PPA variants. Semantic loss eventually becomes apparent in the form of poor recognition of single words. Next, patients show problems with recognition of items around them. Patients also lose knowledge of words that are spelled irregularly, leading to phonetic pronunciations of irregularly spelled words. This is referred to as surface dyslexia (62). They also exhibit regularization of spelling while attempting to spell irregular words, referred to as surface dysgraphia. Despite their semantic loss, patients with svPPA maintain their capacity for repetition and fluency, with normal speech output, intact grammar (aside from some subtle errors related to their poor word knowledge), and intact motoric features of speech.
The right temporal variant of svPPA is also associated with a loss of distinct kinds of conceptual knowledge and an eventual disturbance in language, although the behavioral syndrome is typically the most salient feature of the early disease course (38, 65, 68–70). Patients experience increasingly poor facial recognition coupled with declining semantic knowledge about specific people, starting with individuals to whom they are least frequently exposed (68). Patients experience increasingly poor recognition of emotions in themselves and others, resulting in emotional distance and a cold affect or a childish personality. The typical right temporal behavior syndrome involves hyposexuality, sleep disturbance, and dietary changes, including new dogmatic and faddish preferences. Philosophical intensification is well described in right temporal presentations such as hyperreligiosity (71). Compulsions are common in both forms of svPPA, but people with right temporal svPPA are drawn to words and symbols (manifesting as hypergraphia), whereas those with left temporal svPPA are more often drawn to collection of items (65).
A diagnosis of svPPA requires the previously discussed clinical criteria (Box 2). Supportive features on imaging include asymmetric anterior temporal lobe atrophy on MRI or CT, often most obvious in the inferior temporal gyrus (Figure 2) (45, 72). Predominant anterior temporal hypoperfusion on SPECT or hypometabolism on FDG-PET also may be present (62).
Box 2: DIAGNOSTIC CRITERIA FOR SEMANTIC-VARIANT PRIMARY PROGRESSIVE APHASIA (svPPA)a
I. Clinical diagnosis of svPPA
Both of the following core features must be present:
A.
Impaired confrontation naming
B.
Impaired single-word comprehension
At least three of the following other diagnostic features must be present:
A.
Impaired object knowledge, particularly for low-frequency or low-familiarity items
B.
Surface dyslexia or dysgraphia
C.
Spared repetition
D.
Spared speech production (grammar and motor speech)
II. Imaging-supported svPPA diagnosis
Both of the following criteria must be present:
A.
Clinical diagnosis of svPPA
B.
Imaging must show one or more of the following results:
1.
Predominant anterior temporal lobe atrophy
2.
Predominant anterior temporal hypoperfusion or hypometabolism on SPECT or PET
III. Semantic-variant PPA with definite pathology
Clinical diagnosis (criterion A below) and either criterion B or C must be present:
A.
Clinical diagnosis of semantic variant PPA
B.
Histopathologic evidence of a specific neurodegenerative pathology (e.g. FTLD-tau, FTLD-TDP, AD, other)
C.
Presence of a known pathogenic mutation
___________________
aAdapted from Gorno-Tempini et al. (62)
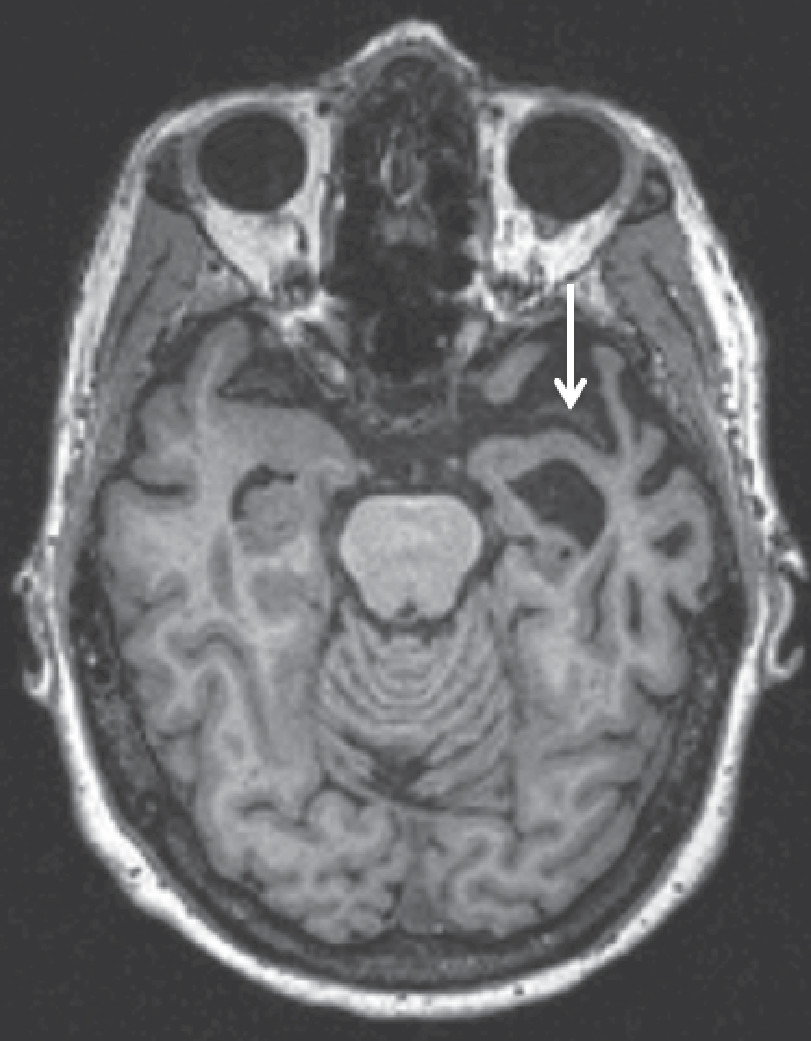
Figure 2. Axial T1-Weighted MRI of a 63-Year-Old Patient with Semantic-Variant Primary Progressive Aphasia (svPPA) in Radiological Orientationa
aAsymmetric left greater than right anterior temporal lobe atrophy noted by an arrow
Nonfluent/Agrammatic-Variant PPA (nfvPPA)
Patients with nfvPPA exhibit dysfunction in frontal regions within the language network, particularly within left inferior frontal and insular cortices (73). The resulting clinical syndrome involves a progressive decline in various aspects of fluency similar to classic Broca’s aphasia syndrome. Patients exhibit decreased ease in confrontation naming, although they retain their knowledge of items and their comprehension of words. A formal diagnosis of nfvPPA requires elements of apraxia of speech or agrammatism, although both are usually present (Box 3) (73). Apraxia of speech may include halting, groping, effortful, and distorted speech with inconsistent motor articulation errors. Sentences become shorter and simpler, and patients may utilize functional nouns and verbs but limit their syntax and verb conjugation. Patients with nfvPPA also frequently experience difficulty in comprehension of grammar and complex syntax, such as passive voice. Repetition is often impaired, similar to conversational speech. A clinical diagnosis of nfvPPA may be supported by MRI or CT brain imaging that reveals predominant left posterior frontoinsular atrophy (Figure 3). Patients may also exhibit hypoperfusion within these regions on SPECT or hypometabolism within these regions on FDG-PET, potentially preceding changes on MRI (74).
Box 3: DIAGNOSTIC FEATURES FOR NONFLUENT/AGRAMMATIC-VARIANT PRIMARY PROGRESSIVE APHASIA (nfvPPA)a
I. Clinical diagnosis of nonfluent/agrammatic variant PPA:
At least one of the following core features must be present:
A.
Agrammatism in language production
B.
Effortful, halting speech with inconsistent speech sound errors and distortions (apraxia of speech)
At least two of the following other features must be present:
A.
Impaired comprehension of syntactically complex sentences
B.
Spared single-word comprehension
C.
Spared object knowledge
II. Imaging-supported nonfluent/agrammatic variant diagnosis
Both of the following criteria must be present:
A.
Clinical diagnosis of nonfluent/agrammatic variant PPA
B.
Imaging must show one or more of the following results:
1.
Predominant left posterior frontoinsular atrophy on MRI or
2.
Predominant left posterior frontoinsular hypoperfusion or hypometabolism on SPECT or PET
III. Nonfluent/agrammatic variant PPA with definite pathology
Clinical diagnosis (criterion A below) and either criterion B or C must be present:
A.
Clinical diagnosis of nonfluent/agrammatic variant PPA
B.
Histopathologic evidence of a specific neurodegenerative pathology (e.g., FTLD-tau, FTLD-TDP, AD, other)
C.
Presence of a known pathogenic mutation
___________________
aAdapted from Gorno-Tempini et al. (62)
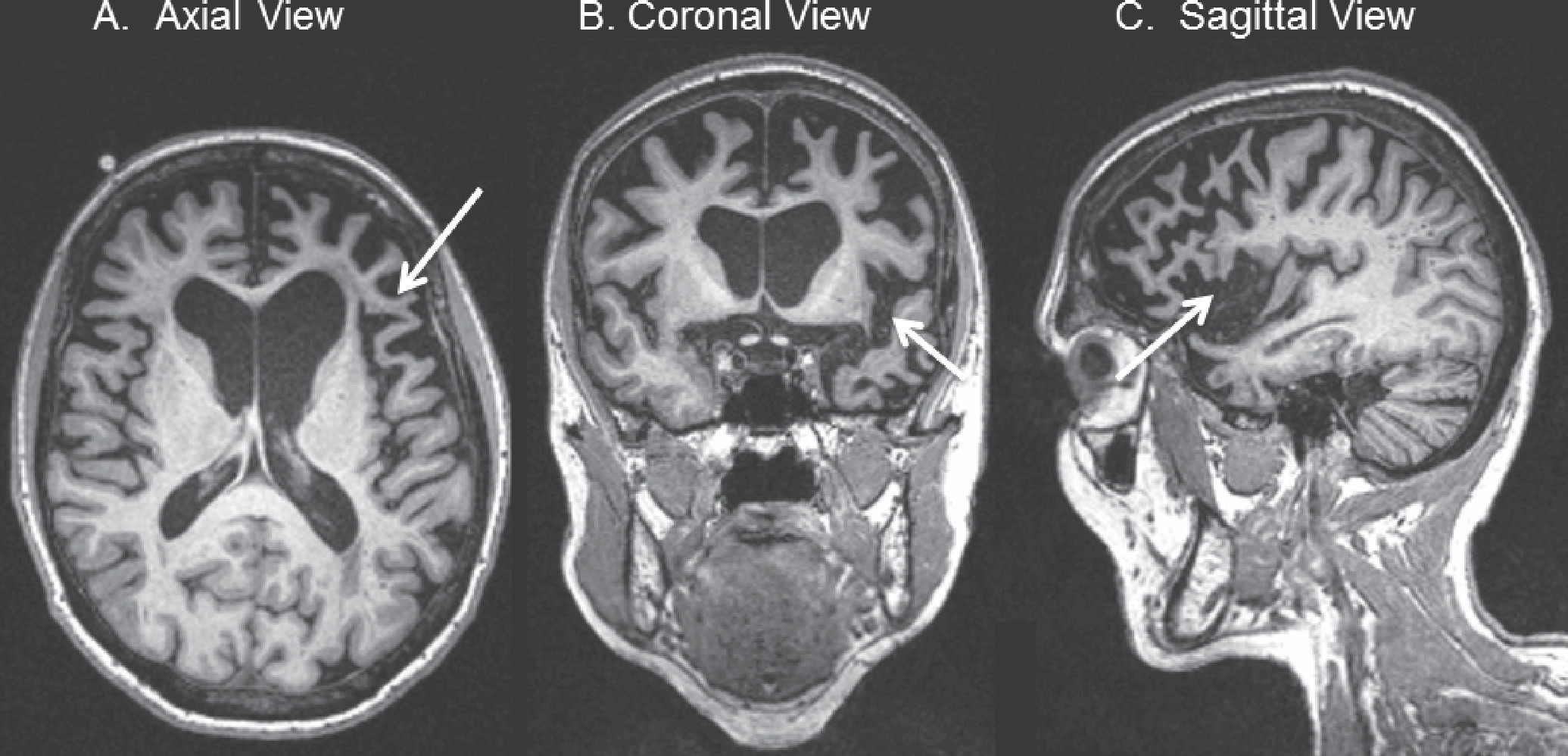
Figure 3. T1-Weighted MRI of a 70-Year-Old Patient With Nonfluent/Agrammatic-Variant Primary Progressive Aphasia (nfvPPA)
A: Axial view in radiological orientation showing asymmetric, left greater than right atrophy of the frontal operculum (arrow). B: Coronal view showing left greater than right atrophy of the anterior perisylvian region (arrow). C: Sagittal view of the left lateral cerebral hemisphere, showing left anterior perisylvian atrophy, including the frontal operculum (arrow).
FTD Spectrum Syndromes Containing Motor Features
FTD Motor Neuron Disease (FTD-MND)
FTD-MND includes patients with clinical features of FTD and amyotrophic lateral sclerosis (ALS). About 15% of patients with clinically confirmed bvFTD then develop MND (19). Additionally, about 30% of patients with ALS have cognitive and behavioral symptoms consistent with bvFTD (75). The behavioral portion of FTD-MND most resembles bvFTD, with a particularly high incidence of early psychosis (76). The motor component of FTD-MND is typical for ALS, involving signs of both upper MND (slowed speech, brisk reflexes, and pathologic reflexes) and lower MND (muscle atrophy and fasciculations) (77). Additionally, FTD-MND is known to have a high burden of early bulbar symptoms (78). As previously mentioned, patients with FTD-MND have much higher rates of positive family histories (including ALS and FTD). Unfortunately, the FTD-MND clinical phenotype confers the shortest mean survival among the FTD syndromes—about 3 years after onset (79).
Corticobasal Syndrome (CBS)
CBS is a progressive clinical phenotype with varied motor and cognitive features resulting from dorsal predominant (often asymmetric) cortical atrophy and a diverse range potential of underlying pathologies. A probable diagnosis of CBS requires at least two of three hallmark motor findings, including limb dystonia, limb myoclonus, and limb rigidity or akinesia (80). A diagnosis also requires at least two of three additional hallmark cortical features, including apraxia (orobuccal or limb apraxia), cortical sensory deficits (involving neglect, agraphesthesia, or astereognosis), or alien limb phenomena. A firm distinction must be drawn between CBS, a clinical syndrome, and corticobasal degeneration (CBD), a pathologic diagnosis that may underlie a variety of clinical syndromes, including CBS, nfvPPA, PSP-S, and bvFTD. CBD accounts for only 35% of clinical CBS, followed by AD (23%), progressive supranuclear palsy (13%), and FTLD with TDP inclusions (13%) (81). MRI allows for relatively good resolution between AD and non-AD pathology underlying CBS. CBS due to underlying AD is associated with atrophy extending into the temporoparietal cortex and precuneus, whereas FTLD pathologies (not limited to CBD) contain atrophy extending into prefrontal cortex, striatum, and brainstem.
Progressive Supranuclear Palsy Syndrome (PSP-S)
The core features of the classic Steele-Richardson-Olszewski PSP-S involve early gait instability with falls (typically backward) and a supranuclear gaze palsy (predominantly impairing the initiation, speed, and eventually the amplitude of vertical saccades) within the first year of disease onset (82). Additional possible supportive features include elements of atypical parkinsonism with axial predominant rigidity, abnormal neck posturing (typically extension), early dysphagia, early dysarthria, and limited responsiveness to l-dopa therapy. The phenotypic overlap between PSP-S and bvFTD is significant. Behavioral features, particularly early apathy and impulsivity, are common, and early cognitive impairment, chiefly involving executive dysfunction, is a supportive feature of the clinical syndrome. Patients also frequently show phenotypic overlap with nfvPPA, with a decrease in verbal fluency as the first manifestation of the illness. Other common, albeit nonspecific, clinical features include a stare, the procerus sign (furrowing of the brow), grasping-utilization of nearby objects, and perseveration (observable as an “applause sign” when they attempt to clap three times) (83). Survival in PSP-S is poor compared with other forms of FTD, with a mean survival of 2.9 years after diagnosis (6).
Clinical descriptions of PSP phenotypes have evolved significantly since the discovery of the classic Steele-Richardson-Olszewski syndrome, which has subsequently been recognized as a narrow portion of the wide clinical spectrum of PSP. A vertically predominant supranuclear gaze palsy, although relatively specific to PSP pathology, may occur late in a patient’s clinical syndrome. Current recognized clinical syndromes associated with PSP underlying pathology include PSP-parkinsonism (which strongly resembles Parkinson’s disease, including moderate l-dopa responsiveness), PSP-pure akinesia with gait freezing (which involves a relatively slow course, severely decreased movement, gait freezing, rigidity, and reduced fine motor tasks), CBS (as described above), and nfvPPA (84). A cerebellar ataxic variant of PSP also has been described, primarily in Japan (85), and some patients with PSP pathology present with typical bvFTD.
Imaging of patients with Steele-Richardson-Olszewski PSP syndrome reveals significant midbrain atrophy of the midbrain tegmentum. This pattern of atrophy may give rise to a so-called hummingbird or penguin sign in a midline sagittal view of the brainstem (Figure 4). Although some authors have explored the validity of this sign as a marker of PSP, its use is controversial. It may, however, be useful in distinguishing PSP from idiopathic Parkinson’s disease, a distinction that can typically be made clinically (86).
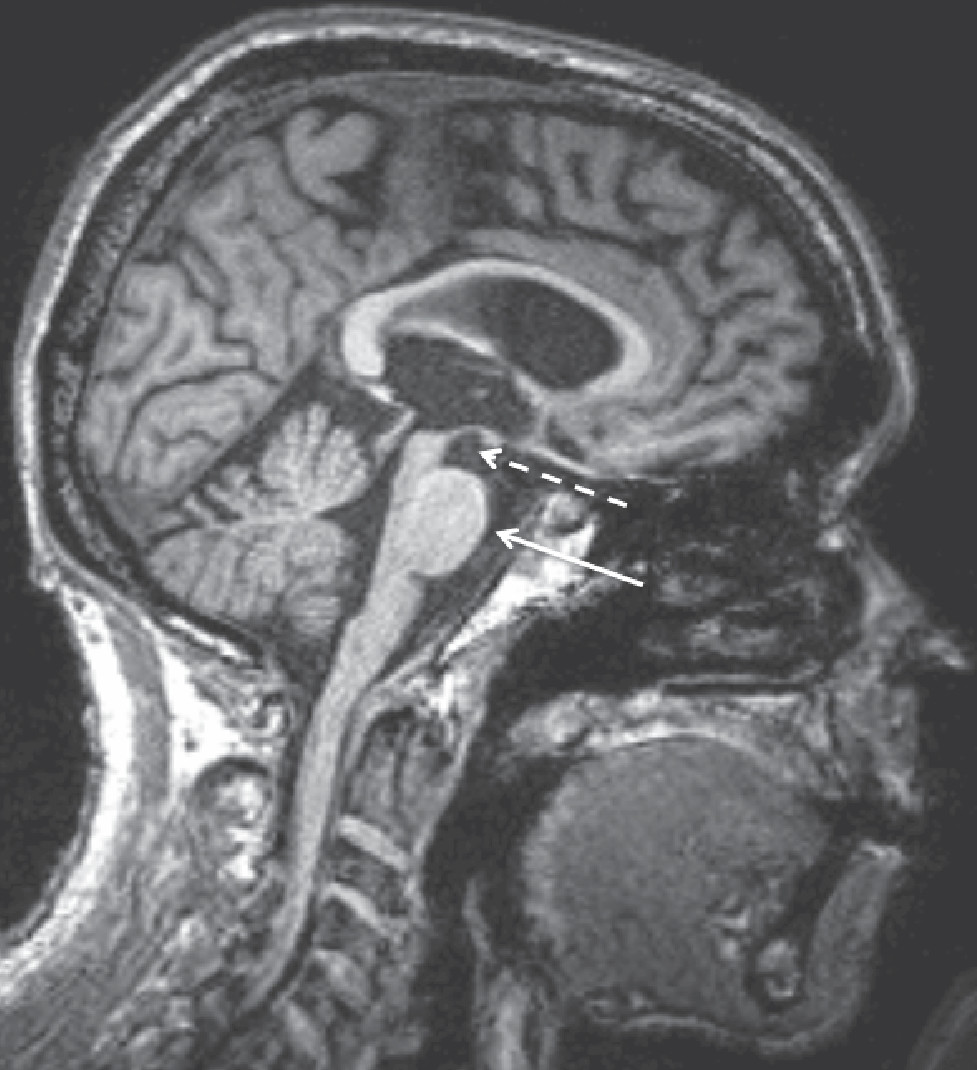
Figure 4. Sagittal T1-Weighted MRI of a 71-Year-Old Patient With Progressive Supranuclear Palsy (PSP)a
aImage shows “hummingbird sign,” including an atrophic midbrain tegmentum (dashed arrow) resembling a hummingbird’s head and a relatively preserved basis pontis (solid arrow) that resembles the hummingbird’s belly.
FTLD Clinical Pathological Correlates
All cases of FTLD share common pathologic findings, including synapse loss, gliosis, neuronal loss, and gross atrophy within the frontal and anterior temporal lobes (1). Further distinctions between FTLD cases may be made with attention to a variety of intracellular protein inclusions within affected brain parenchyma (1). Historical descriptions of FTLD focused on a particular subset of pathology now known as Pick’s disease, which chiefly involves aggregation of interneuronal and glial aggregates containing a specific isoform of hyperphosphorylated tau (87, 88). As descriptions evolved, FTLD was increasingly categorized into two roughly equal subcategories: cases with intracellular tau inclusions called FTLD-tau (of which Pick’s disease was one variety) and cases containing intracellular ubiquinated inclusions called FTLD-U. In 2006, the majority of ubiquinated inclusions among FTLD-U cases were found to comprise mainly TDP-43 (transactive response DNA binding protein 43 kDa), a DNA and RNA binding protein found in the nucleus of normal cells (89). Current FTLD nomenclature distinguishes between a diverse variety of tau and TDP-43 pathology subcategories. Roughly 90%−95% of known FTLD cases can be divided evenly between tau and TDP-43 subpathologies (90). Most of the remaining 5%−10% of FTLD cases contain intracellular inclusions of FUS (fused-in sarcoma), and only a small minority of cases represents alternate pathologies. Although there is not a strong correlation between the various FTLD subtypes and clinical syndromes, there are various trends in anatomic distribution and associated clinical symptomology that may hint at an underlying pathology in living individuals (Table 1).
| FTD Organized by Clinical Syndromes | |
|---|---|
| FTD Clinical Syndrome | Typically Associated FTLD Pathology |
| bvFTD | Tau (3R > 4R) ≈ TDP-43 (type A ≥ type B > type D) > (Alzheimer’s pathology) > FUS |
| svPPA | TDP-43 type C > 3R tau |
| nfvPPA | 4R tau (CBD and PSP) > 3R tau and TDP-43 type A |
| FTD-MND | TDP-43 type B > TDP-43 type A > FUS |
| CBS | 4R tau (CBD) > (Alzheimer’s pathology) > PSP, TDP-43 > 3R tau |
| PSP-S | 4R tau (PSP > CBD) |
| FTD Organized by Underlying Pathologies | |
| FTLD Pathology Subtypes (and sites of rare mutations) | Typical FTD Clinical Syndrome |
| FTLD-tau: (MAPT) | |
| 3R tau (Pick’s disease): | bvFTD > nfvPPA, svPPA, CBS |
| 4R tau: | |
| CBD | Motor dysexecutive (including CBS) > PSP-S, nfvPPA, bvFTD |
| PSP | PSP-S > CBS, nfvPPA, bvFTD |
| FTLD-TDP | |
| Type A: (GRN) | bvFTD > CBS, nfvPPA > FTD-MND |
| Type B: (C9ORF72) | bvFTD, FTD-MND, ALS |
| Type C | svPPA |
| Type D (always VCP mutation) | bvFTD, ALS, Paget’s disease of bone, inclusion body myositis |
| FTLD-FUS (FUS gene) | |
| aFTLD-U, BIBD, NIFID | bvFTD if sporadic versus ALS if genetic |
a
FTD, frontotemporal dementia; bvFTD, behavioral-variant FTD; svPPA, semantic-variant primary progressive aphasia; nfvPPA, nonfluent/agrammatic-variant PPA; FTD-MND, FTD motor neuron disease; CBS, corticobasal syndrome; CBD, corticobasal degeneration; PSP, progressive supranuclear palsy; ALS, amyotrophic lateral sclerosis; aFTLD-U, atypical FTLD with ubiquitin inclusions; BIBD, basophilic inclusion body disease; NIFID, neuronal intermediate filament inclusion disease.
FTLD-Tau
Tau protein is encoded by the MAPT gene on chromosome 17 and is involved in a variety of cell processes, including stabilization of microtubules, promotion of microtubule assembly, and regulation of axonal transport (91). Alternative splicing of premRA gives rise to at least six tau isoforms (92). Half of these isoforms contain three copies of a repeated microtubule-binding domain (3R tau), and the remaining isoforms contain four copies (4R tau). 3R and 4R tau may be found in roughly equal proportions within the neurons of healthy individuals and the tau tangles associated with AD. The various subcategories of FTLD-tau may, however, be distinguished via the predominance of either hyperphosphorylated 3R or 4R tau (with the expectation of rare FTD presentations related to mutations in the MAPT gene itself, which tend to have a more balanced ratio).
3R tauopathy.
Pick’s disease includes FTLD pathology with intracellular hyperphosphorylated tangles containing chiefly 3R tau. These aggregates typically start in frontotemporal limbic structures early in disease (93). They later involve the basal ganglia, locus coeruleus, and raphe nuclei followed by primary motor cortex. The pattern of progression may include profound frontotemporal atrophy (with so-called knife gyri) and relative sparing of parietal lobes, globus palidus, and thalamus. Pick’s disease most commonly results in bvFTD and comprises the largest proportion of bvFTD cases with underlying tau (93). A significant minority may alternatively present with nfvPPA and CBS phenotypes, or less commonly svPPA (81, 93, 94). Pick’s disease is typically associated with a later disease onset (mean onset of 57 years) and a longer duration (mean duration of 8–9 years) compared with other FTLD subtypes (79, 93).
4R tauopathies.
Most 4R tau pathology results in the pathological diagnoses of CBD and pathologically defined PSP.
CBD pathology involves asymmetric frontoparietal atrophy grossly. Microscopic features of CBD include extensive neuronal loss, ballooned achromatic neurons (which are less common in PSP), and astrocytic plaques in focal cortices with atrophy (95). Intraneuronal pathology contains 4R tau in filamentous, fine, wispy inclusions throughout cerebral gray and white matter (though they are not excluded from the basal ganglia and brainstem) (95). CBD pathology typically gives rise to a dorsal predominant pattern of frontal atrophy, with relative temporal lobe sparing and a diverse range of clinical phenotypes. CBD most commonly results in a dysexecutive motor phenotype, which includes cases that meet the more narrow clinical criteria for CBS and occasionally cases with a PSP clinical features (81). CBD is also the most frequent cause of nfvPPA and may give rise to a typical bvFTD syndrome (94).
PSP pathology most prominently involves the basal ganglia, diencephalon, and brainstem (95). Intraneuronal PSP pathology contains dense, compact, filamentous aggregates in globose neurofibrillary tangles containing 4R tau. Glial pathology includes tufted astrocytic lesions in the motor cortex and striatum. PSP pathology typically gives rise to the diversity of syndromes within the clinical PSP spectrum described earlier. However, PSP pathology is also the second most common cause of nfvPPA, and it may give rise to typical CBS and bvFTD clinical presentations (94, 96). Although there may be significant clinical overlap between presentations of CBS and PSP pathology, supranuclear gaze palsy is relatively specific for underlying PSP.
Other 4R tauopathies include globular glial tauopathies (GGT) and argyrophilic grain disease (AGD). GGT is a rare pathology that involves tau-positive globular oligodendroglial inclusions and gives rise to the wide spectrum of FTD clinical syndromes with prominent white matter change on MRI (97, 98). AGD pathology is largely restricted to the medial temporal lobes and limbic regions and is generally thought to give a slow progressive amnestic phenotype similar to AD (99).
FTLD-TDP
Interneuronal aggregates of TDP-43 are found in the majority of patients with ALS and a sizeable proportion of FTD patients (100). Current naming conventions organize TDP pathology into four subtypes, types A through D, based on immunohistochemical staining of TDP-43 containing neuronal cytoplasmic inclusions (NCIs) and dystrophic neurites of affected neurons (101, 102).
Type A.
Pathology is defined by moderate to numerous NCIs and short dystrophic neurites predominantly in upper cortical layers II or III. TDP-43 type A pathology accounts for about half of all FTLD-TDP. Although cases are typically sporadic, this pathology often is associated with mutations in the GRN gene and sometimes with pathologic expansion of the C9ORF72 gene (58, 102). The most common clinical presentation of TDP-43 type A is bvFTD, but CBS and nfvPPA presentations occur frequently, and FTD-MND presentation may also occur (81, 94, 101).
Type B.
Pathology is defined by numerous NCIs across all cortical layers but relatively few dystrophic neurites. This pathology represents the largest proportion of TDP pathology that gives rise to the bvFTD and FTD-MND clinical syndromes. Most cases of TDP-43 type B pathology are sporadic, but some cases are commonly associated with the pathologic expansion of the C9ORF72 gene and rarely with mutations within the gene coding for TDP-43 (58).
Type C.
Pathology is defined by long dystrophic neurites and relatively few NCIs, mostly in upper cortical layers. The majority of TDP-43 type C cases are associated with asymmetric temporal lobe atrophy correlating with an svPPA clinical syndrome, and this pathology underlies most svPPA cases (103). Unlike other TDP-43 pathology, type C is almost never familial (67).
Type D.
Pathology is defined by numerous lentiform intranuclear inclusions. This pathologic subtype is associated with rare mutations such as the valosin-containing protein (VCP) gene, which give rise to a set of diseases including FTD, ALS, Paget’s disease of bone, and inclusion body myositis (104).
FTLD-FUS
The majority of rare FTLD cases lacking tau and TDP-43 pathology involve interneuronal aggregates of FUS protein, a DNA binding protein with homology to TDP-43 (105). The most common form of FTLD-FUS is atypical FTLD with ubiquitin inclusions (aFTLD-U), but other varieties include basophilic inclusion body disease (BIBD) and neuronal intermediate filament inclusion disease (NIFID) (102, 106, 107). Genetic mutations in the FUS gene are strongly associated with ALS but can also lead to bvFTD presentations. Sporadic FUS pathology is strongly associated with a distinct bvFTD presentation with an early age of onset (22–46 years), obsessiveness, compulsive behavior, social withdrawal, hyperorality (often with pica), stimulus-boundedness, and the unique feature of severe caudate atrophy on MRI (108). FUS pathology may also give rise to FTD-MND.
Alzheimer’s Disease (AD) Pathology
AD pathology is disguised by the presence of extracellular amyloid beta–containing plaques, as well as intracellular deposits and interneuronal neurofibrillary tangles containing hyperphosphorylated tau (109). Although many FTLD presentations contain a predominance of 3R or 4R tau isoforms, these isoforms occur in roughly equal proportions within the tau tangles found in AD. AD pathology is technically exclusionary for a research diagnosis of bvFTD. AD pathology may, however, result in clinical syndromes that meet diagnostic criteria for bvFTD, PPA, and CBS (49, 62, 81). The clinical differentiation between FTD and frontal AD is detailed earlier in our discussion of the diagnosis of bvFTD.
Genes of Risk in FTLD
Most FTD occurs sporadically, but up to 20% of cases have a known familial cause, and up to 40% of cases have a strong family history without a clear pattern of inheritance (6, 8, 11, 12). To date, there are three sites of autosomal-dominant mutations (C9ORF72, MAPT, and GRN) that account for the most common known causes of familial FTLD.
Chromosome 9 Open Reading Frame 72 (C9ORF72)
Pathological expansion of the C9ORF72 gene is the most common genetic cause of familial FTD and familial ALS, accounting for 11.7% and 23.5% of familial cases, respectively (110). Healthy individuals typically have less than 30 hexanucleotide (GGGGCC) repeats located between noncoding C9ORF72 exons 1a and 1b, whereas patients with 700–1,600 repeats experience nearly 100% penetrance with either FTD, ALS, or an FTD-MND syndrome. Pathologic expansion of C9ORF72 is known to impair nucleocytoplasmic transport, although the precise mechanism for neurotoxicity has yet to be firmly established (111). The majority of C9ORF72 cases develop TDP-43 type B pathology, although a minority of cases result in TDP-43 type A pathology (58). Hexanucleotide expansions also undergo non-ATG–initiated translation, resulting in aggregates of dipeptide-repeat proteins within the cerebellum, hippocampus, and frontotemporal neocortex (112). Patients with pathologic expansion of C9ORF72 are distinctly more likely than other FTD gene carriers to exhibit psychotic features. In one cohort of 38 carriers of C9ORF72 mutations, 38% of patients presented with psychosis, and an additional 28% exhibited paranoia, delusion, or irrational thinking (58). Interestingly, C9ORF72 mutation carriers are more likely than other FTD patients to exhibit surprising social warmth and appropriateness at presentation. FTD-MND is a typical clinical phenotype for C9ORF72 mutation carriers, although carriers commonly present with pure FTD or pure ALS syndromes. There is also literature to suggest rare presentations with CBS, PSP, PPA, and a Huntington’s disease-like syndrome (113). Unique radiographic features to C9ORF72 mutations carriers include a relatively high burden of cerebellar and thalamic atrophy compared with other patients with FTD (114, 115).
Microtubule-Associated Protein Tau (MAPT)
The MAPT gene, located on chromosome 17, codes for tau protein and is the site of numerous autosomal-dominant mutations that give rise to FTLD. The cluster of familial syndromes associated with the MAPT mutations are known as FTD and parkinsonism linked to chromosome 17 (FTDP-17) (116). Carriers of the MAPT gene mutation tend to have a younger age of onset (commonly under 50) (61). Patients present with diverse clinical syndromes, including bvFTD, PPA, CBS, and PSP (117). The most commonly described clinical phenotype in recent literature involves mixed features of bvFTD and svPPA, typically with early behavioral disinhibition and temporal lobe atrophy leading to semantic loss (61). About half of patients also have parkinsonian features. MND and early apathy are not common features to FTDP-17.
Progranulin (GRN)
Progranulin is a glycoprotein with many known functions, including regulation of inflammation, stress response, and apoptosis (118). Heterozygous loss of the GRN gene leads to an increased risk of FTLD pathology with TDP-43 type A inclusions, whereas homozygous loss of function leads to neuronal ceroid lipofuscinosis (119). Patients with GRN deficiency tend to have a later onset (mean of 59 years) and a long disease duration (a mean of 9 years) compared with other FTD mutation carriers (61). FTD in GRN mutation carriers has distinct radiographic features on MRI, typically involving asymmetric frontotemporal and parietal atrophy. Patients also have T2 hyperintense confluent white matter disease affecting the periventricular subcortical white matter and U-fibers (120). Although GRN deficiency most commonly results in bvFTD (typically with early apathy), it also results in nfvPPA and CBS more frequently than other FTD mutations (61, 121). GRN mutation carriers occasionally experience psychosis, although at a lower frequency than carriers of C9ORF72 mutations (61).
Other Mutations
Several additional mutations have been described with autosomal-dominant patterns of inheritance, although these genes are rare causes of FTD. Interestingly, many of these mutations frequently result in ALS. Mutations in chromatin-modifying protein 2B (CHMP2B) may, for instance, give rise to FTD (with vacuolar cortical pathology rather than prominent protein inclusions) or ALS clinical phenotypes (122, 123). Mutations in tank binding kinase 1 (TBK1) also result a clinical spectrum including FTD, ALS, and FTD-MND (124). Mutations in FUS and TDP-43 genes almost exclusively give rise to ALS, although a few clinical presentations with FTD have been described (125, 126). Additionally, mutations within VCP give rise to FTLD (with TDP-43 type D pathology), inclusion body myositis, Paget’s disease of bone, and familial ALS (104).
Treatment of FTD
Nonpharmacologic Interventions
Nonpharmacologic interventions should be considered the first line of treatment for patients with FTD. Patients must establish care with a primary care provider immediately so that future care may be better coordinated. The topics of driving and home safety should be broached on the first visit. Driving safety should be a serious concern if patients exhibit aggression or impulsivity, or if caregivers report marginal or unsafe driving (127). Other red flags for driver safety include recent car accidents, recent traffic citations, and deliberate restriction of mileage. Optimization of home safety should include removal of firearms or other dangerous implements from the home. Early topics of discussion should also include the loss of financial independence and decision making, power of attorney arrangements, application for disability, early retirement, and the potential need for supervision.
Caregiver education and environmental intervention are important tools in the management of bvFTD. Insight into the underlying nature of FTD allows caregivers to view neuropsychiatric features as symptoms of a disease rather than as volitional actions. Caregivers should be encouraged to pursue strategies involving distraction, redirection, and simplification of complex decisions rather than confrontation and rational debate. Social engagement is important, but patients should avoid overstimulation in favor of an ordered and structured environment. Patients benefit from restriction of access to items that may incite unwanted behaviors (e.g., car keys or excess food). Caregiver support and respite should also be considered early in management, given that the risk of caregiver burnout is high in cases of bvFTD. Resources may be found via organizations such as the Family Caregiver Alliance (www.caregiver.org) and the Frontotemporal Dementia Caregiver Support Center (ftdsupport.com).
Nonbehavioral interventions are also pertinent in FTD. Consultation with a speech pathologist may help patients to cope in the earliest stages of PPA. Many patients with bvFTD eventually pass away from aspiration pneumonia, so a formal evaluation of swallowing is also important. Exercise, particularly under the direction of a physical therapist and personal trainers, may allow patients to maintain their balance and mobility for longer, even in the context of parkinsonism. Regular exercise may have the added benefit of improving mood, behavior, functional independence, and vascular health (128, 129). Vascular health (and the risk of concurrent vascular dementia) also may be aided with a Mediterranean diet rich in fish, legumes, fruits, vegetables, and unsaturated fatty acids, and low in meat and dairy products.
Pharmacologic Interventions
To date, there are no FDA-approved therapies for the treatment of FTD. However, several medications have shown varying degrees of modest efficacy in managing the symptoms of FTD. Generally, titrations of pharmacotherapy should be done slowly and judiciously in all cases of FTD, with careful awareness of potential behavioral, extrapyramidal, and cognitive side effects.
Various medications may be considered to optimize the availability and function of several neurotransmitters in patients with bvFTD. Serotonin is an important neurotransmitter in frontal subcortical pathways, and loss of serotonergic binding is documented in bvFTD (130, 131). Early case reports and case series studies using various selective serotonin reuptake inhibitors (including fluoxetine, sertraline, and paroxetine) have some efficacy in treating compulsion, hyperorality, depression, inappropriate sexual behavior, and disinhibition of patients with bvFTD (132, 133). Reduction in behavioral symptoms (including disinhibition and irritability) and caregiver stress have also been observed in open-label trials with citalopram and paroxetine, although paroxetine efficacy was not replicated in a subsequent double-blind randomized controlled trial (134–136). Trazodone has been shown to improve neuropsychiatric symptoms of bvFTD, which includes assessment of agitation, irritability, eating disorders, and depressive symptoms in a randomized placebo-controlled trial (137). Patients with FTD may also have a deficit in dopaminergic function, but there is limited information that augmentation of this deficit improves FTD symptoms (138). A small trial of methylphenidate suggested efficacy in managing risk-taking behavior (139). Another small trial comparing dextro-methamphetamine to quetiapine documented relatively less apathy and disinhibition with stimulant use (140). Overall, due to the paucity of information and the risk of delirium, stimulant use is generally discouraged for patients with FTD. The neuropeptide oxytocin has been suggested as a potential treatment for empathy loss in FTD and has been associated with improvement, although it has not been involved in wide clinical use (141).
The typical pharmacologic interventions used in Alzheimer’s disease are unlikely to clinically benefit patients with FTD who are not known to have a deficit in cholinergic function (142). A placebo-controlled trial of galantamine showed no significant improvement in behavior or language symptoms among patients with bvFTD and PPA, and a slight trend of efficacy (in global severity and language) in the PPA subgroup may have been explained by the inclusion of lvPPA, an Alzheimer’s variant, in the study cohort (143). Additionally, although rivastigmine dosing has been associated with modest behavioral improvement, an open-label trial of donepazil documented clear worsening and disinhibition in patients with bvFTD (144, 145). Memantine is generally well tolerated but has shown no benefit in cognition or behavior in double-blind placebo-controlled studies involving patients with bvFTD (146, 147).
Antipsychotics and mood stabilizers are occasionally used to treat psychosis and agitation of patients with FTD, although there is inherent risk and a paucity of evidence associated with their use. Risperidone and olanzapine have some documented utility in treating neuropsychiatric symptoms in dementia compared with other antipsychotics, but their efficacy is still modest (148). In the case of patients with parkinsonism, low-affinity D2 blockers such as quetiapine are preferable to other antipsychotics. Unfortunately, all antipsychotics increase the risk of cardiovascular events and mortality of elderly patients with dementia. The mood stabilizer and antiepileptic valproate has been used to calm patients with FTD without illicit parkinsonism, but documentation of its use has been scant (149). A small trial with topiramate documented efficacy in limiting alcohol abuse by patients with FTD, but no other behavioral benefits could be associated with its use (150).
Parkinsonism associated with FTD is rarely responsive to l-dopa therapy, although some patients may experience a modest response. A trial of levodopa/carbidopa is warranted in cases with prominent parkinsonism, although care must be taken to avoid dystonia, dyskinesia, and visual hallucination.
Potential for Future Interventions
Although there are yet no disease-modifying interventions for FTD, growing recognition of pathological subtypes and underlying mechanisms of FTLD has generated enthusiasm for a variety of tailored interventions for various FTD categories. One potential target for intervention is the toxic gain of function for phosphorylated tau in FTLD-tau variants. Protein kinase inhibitors to limit tau phosphorylation and antitau antibodies are being brought to trial for treatment of 4R tauopathies (151). Toxic gain of function has also been discussed as a potential target for antisense oligonucleotide therapies in C9ORF72 mutation carriers (152). Antisense oligonucleotides also present the potential for editing the ratio of 4R and 3R tau in vivo (153). Toxic loss of tau function is another proposed mechanism for drugs that may stabilize microtubules. Additionally, augmentation of haploinsufficiency is a potential target for patients with GRN deficiency (151). Finally, based on case report information, there is ongoing interest in immunotherapies for treatment of svPPA (154).
References
1.
Brun A, Liu X, Erikson C: Synapse loss and gliosis in the molecular layer of the cerebral cortex in Alzheimer’s disease and in frontal lobe degeneration. Neurodegeneration 1995; 4:171–177
2.
Mercy L, Hodges JR, Dawson K, et al: Incidence of early-onset dementias in Cambridgeshire, United Kingdom. Neurology 2008; 71:1496–1499
3.
Ratnavalli E, Brayne C, Dawson K, et al: The prevalence of frontotemporal dementia. Neurology 2002; 58:1615–1621
4.
Johnson JK, Diehl J, Mendez MF, et al: Frontotemporal lobar degeneration: demographic characteristics of 353 patients. Arch Neurol 2005; 62:925–930
5.
Knopman DS, Roberts RO: Estimating the number of persons with frontotemporal lobar degeneration in the US population. J Mol Neurosci 2011; 45:330–335
6.
Coyle-Gilchrist ITS, Dick KM, Patterson K, et al: Prevalence, characteristics, and survival of frontotemporal lobar degeneration syndromes. Neurology 2016; 86:1736–1743
7.
Onyike CU, Diehl-Schmid J. The epidemiology of frontotemporal dementia. Int Rev Psychiatry 2013; 25:130–137.
8.
Rosso SM, Donker Kaat L, Baks T, et al: Frontotemporal dementia in the Netherlands: patient characteristics and prevalence estimates from a population-based study. Brain 2003; 126:2016–2022
9.
Ikejima C, Yasuno F, Mizukami K, et al: Prevalence and causes of early-onset dementia in Japan: a population-based study. Stroke 2009; 40:2709–2714
10.
Borroni B, Alberici A, Grassi M, et al: Is frontotemporal lobar degeneration a rare disorder? Evidence from a preliminary study in Brescia County, Italy. J Alzheimers Dis 2010; 19:111–116
11.
Goldman JS, Farmer JM, Wood EM, et al: Comparison of family histories in FTLD subtypes and related tauopathies. Neurology 2005; 65:1817–1819
12.
Le Ber I, Guedj E, Gabelle A, et al:Demographic, neurological and behavioural characteristics and brain perfusion SPECT in frontal variant of frontotemporal dementia. Brain 2006; 129:3051–3065
13.
Roberson ED, Hesse JH, Rose KD, et al: Frontotemporal dementia progresses to death faster than Alzheimer disease. Neurology 2005; 65:719–725
14.
Kertesz A, McMonagle P, Blair M, et al: The evolution and pathology of frontotemporal dementia. Brain 2005; 128:1996–2005
15.
Woollacott IO, Rohrer JD: The clinical spectrum of sporadic and familial forms of frontotemporal dementia. J Neurochem (Epub May 3, 2016)
16.
Schroeter ML, Raczka K, Neumann J, et al: Neural networks in frontotemporal dementia—a meta-analysis. Neurobiol Aging 2008; 29:418–426
17.
Seeley WW, Crawford RK, Zhou J, et al: Neurodegenerative diseases target large-scale human brain networks. Neuron 2009; 62:42–52
18.
Rademakers R, Neumann M, Mackenzie IR: Advances in understanding the molecular basis of frontotemporal dementia. Nat Rev Neurol 2012; 8:423–434
19.
Rascovsky K, Hodges JR, Knopman D, et al: Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 2011; 134:2456–2477
20.
Rosen HJ, Allison SC, Schauer GF, et al: Neuroanatomical correlates of behavioural disorders in dementia. Brain 2005; 128:2612–2625
21.
Seeley WW, Crawford R, Rascovsky K, et al: Frontal paralimbic network atrophy in very mild behavioral variant frontotemporal dementia. Arch Neurol 2008; 65:249–255
22.
Mendez MF, Lauterbach EC, Sampson SM: An evidence-based review of the psychopathology of frontotemporal dementia: a report of the ANPA Committee on Research. J Neuropsychiatry Clin Neurosci 2008; 20:130–149
23.
Massimo L, Powers C, Moore P, et al: Neuroanatomy of apathy and disinhibition in frontotemporal lobar degeneration. Dement Geriatr Cogn Disord 2009; 27:96–104
24.
Holroyd CB, Yeung N: Motivation of extended behaviors by anterior cingulate cortex. Trends Cogn Sci 2012; 16:122–128
25.
Sollberger M, Rosen HJ, Shany-Ur T, et al: Neural substrates of socioemotional self-awareness in neurodegenerative disease. Brain Behav 2014; 4:201–214
26.
Tranel D, Bechara A, Denburg NL: Asymmetric functional roles of right and left ventromedial prefrontal cortices in social conduct, decision-making, and emotional processing. Cortex 2002; 38:589–612
27.
Sturm VE, Sollberger M, Seeley WW, Rankin KP, Ascher EA, Rosen HJ, et al: Role of right pregenual anterior cingulate cortex in self-conscious emotional reactivity. Soc Cogn Affect Neurosci 2013; 8:468–474
28.
Eckart JA, Sturm VE, Miller BL, et al: Diminished disgust reactivity in behavioral variant frontotemporal dementia. Neuropsychologia 2012; 50:786–790
29.
Wright P, He G, Shapira NA, et al: Disgust and the insula: fMRI responses to pictures of mutilation and contamination. Neuroreport 2004; 15:2347–2351
30.
Ahmed RM, Kaizik C, Irish M, et al: Characterizing sexual behavior in frontotemporal dementia. J Alzheimers Dis 2015; 46:677–686
31.
Granadillo ED, Mendez MF: Pathological joking or Witzelsucht revisited. J Neuropsychiatry Clin Neurosci 2016; 28:162–167
32.
Williamson C, Alcantar O, Rothlind J, et al: Standardised measurement of self-awareness deficits in FTD and AD. J Neurol Neurosurg Psychiatry 2010; 81:140–145
33.
Seeley WW: Anterior insula degeneration in frontotemporal dementia. Brain Struct Funct 2010; 214:465–475
34.
Rankin KP, Gorno-Tempini ML, Allison SC, et al: Structural anatomy of empathy in neurodegenerative disease. Brain 2006; 129:2945–2956
35.
Perry DC, Whitwell JL, Boeve BF, et al: Voxel-based morphometry in patients with obsessive-compulsive behaviors in behavioral variant frontotemporal dementia. Eur J Neurol 2012; 19:911–917
36.
Woolley JD, Gorno-Tempini ML, Seeley WW, et al: Binge eating is associated with right orbitofrontal-insular-striatal atrophy in frontotemporal dementia. Neurology 2007; 69:1424–1433
37.
Piguet O: Eating disturbance in behavioural-variant frontotemporal dementia. J Mol Neurosci 2011; 45:589–593
38.
Henry ML, Wilson SM, Ogar JM, et al: Neuropsychological, behavioral, and anatomical evolution in right temporal variant frontotemporal dementia: a longitudinal and post-mortem single case analysis. Neurocase 2014; 20:100–109
39.
Kramer JH, Jurik J, Sha SJ, et al: Distinctive neuropsychological patterns in frontotemporal dementia, semantic dementia, and Alzheimer disease. Cogn Behav Neurol 2003; 16:211–218
40.
Huey ED, Goveia EN, Paviol S, et al: Executive dysfunction in frontotemporal dementia and corticobasal syndrome. Neurology 2009; 72:453–459
41.
Torralva T, Roca M, Gleichgerrcht E, et al: A neuropsychological battery to detect specific executive and social cognitive impairments in early frontotemporal dementia. Brain 2009; 132:1299–1309
42.
Hornberger M, Piguet O, Graham AJ, et al: How preserved is episodic memory in behavioral variant frontotemporal dementia? Neurology 2010; 74:472–479
43.
Rosen HJ, Narvaez JM, Hallam B, et al: Neuropsychological and functional measures of severity in Alzheimer disease, frontotemporal dementia, and semantic dementia. Alzheimer Dis Assoc Disord 2004; 18:202–207
44.
Perry RJ, Graham A, Williams G, et al: Patterns of frontal lobe atrophy in frontotemporal dementia: a volumetric MRI study. Dement Geriatr Cogn Disord 2006; 22:278–287
45.
Rosen HJ, Gorno-Tempini ML, Goldman WP, et al: Patterns of brain atrophy in frontotemporal dementia and semantic dementia. Neurology 2002; 58:198–208
46.
Seeley WW: Selective functional, regional, and neuronal vulnerability in frontotemporal dementia. Curr Opin Neurol 2008; 21:701–707
47.
Mendez MF, Shapira JS, McMurtray A, et al: Accuracy of the clinical evaluation for frontotemporal dementia. Arch Neurol 2007; 64:830–835
48.
Foster NL, Heidebrink JL, Clark CM, et al: FDG-PET improves accuracy in distinguishing frontotemporal dementia and Alzheimer’s disease. Brain 2007; 130:2616–2635
49.
Ossenkoppele R, Pijnenburg YA, Perry DC, et al: The behavioural/dysexecutive variant of Alzheimer’s disease: clinical, neuroimaging and pathological features. Brain 2015; 138:2732–2749
50.
Ewers M, Mattsson N, Minthon L, et al: CSF biomarkers for the differential diagnosis of Alzheimer’s disease: A large-scale international multicenter study. Alzheimers Dement 2015; 11:1306–1315
51.
McKhann GM, Knopman DS, Chertkow H, et al: The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 2011; 7:263–269
52.
Ossenkoppele R, Jansen WJ, Rabinovici GD, et al: Prevalence of amyloid PET positivity in dementia syndromes: a meta-analysis. JAMA 2015; 313:1939–1949
53.
Mendez MF, Shapira JS, Woods RJ, et al: Psychotic symptoms in frontotemporal dementia: prevalence and review. Dement Geriatr Cogn Disord 2008; 25:206–211
54.
Berg D, Postuma RB, Adler CH, et al: MDS research criteria for prodromal Parkinson’s disease. Mov Disord 2015; 30:1600–1611
55.
Rascovsky K, Grossman M: Clinical diagnostic criteria and classification controversies in frontotemporal lobar degeneration. Int Rev Psychiatry 2013; 25:145–158
56.
Khan BK, Yokoyama JS, Takada LT, et al: Atypical, slowly progressive behavioural variant frontotemporal dementia associated with C9ORF72 hexanucleotide expansion. J Neurol Neurosurg Psychiatry 2012; 83:358–364
57.
Velakoulis D, Walterfang M, Mocellin R, et al: Frontotemporal dementia presenting as schizophrenia-like psychosis in young people: clinicopathological series and review of cases. Br J Psychiatry 2009; 194:298–305
58.
Snowden JS, Rollinson S, Thompson JC, et al: Distinct clinical and pathological characteristics of frontotemporal dementia associated with C9ORF72 mutations. Brain 2012; 135:693–708
59.
Lanata SC, Miller BL: The behavioural variant frontotemporal dementia (bvFTD) syndrome in psychiatry. J Neurol Neurosurg Psychiatry 2016; 87:501–511
60.
Galimberti D, Dell’Osso B, Fenoglio C, et al: Progranulin gene variability and plasma levels in bipolar disorder and schizophrenia. PLoS One 2012; 7:e32164
61.
Snowden JS, Adams J, Harris J, et al: Distinct clinical and pathological phenotypes in frontotemporal dementia associated with MAPT, PGRN and C9orf72 mutations. Amyotroph Lateral Scler Frontotemporal Degener 2015; 16:497–505
62.
Gorno-Tempini ML, Hillis AE, Weintraub S, et al: Classification of primary progressive aphasia and its variants. Neurology 2011; 76:1006–1014
63.
Mesulam M, Wicklund A, Johnson N, et al: Alzheimer and frontotemporal pathology in subsets of primary progressive aphasia. Ann Neurol 2008; 63:709–719
64.
Guo CC, Gorno-Tempini ML, Gesierich B, et al: Anterior temporal lobe degeneration produces widespread network-driven dysfunction. Brain 2013; 136:2979–2991
65.
Seeley WW, Bauer AM, Miller BL, et al: The natural history of temporal variant frontotemporal dementia. Neurology 2005; 64:1384–1390
66.
Miller ZA, Mandelli ML, Rankin KP, et al: Handedness and language learning disability differentially distribute in progressive aphasia variants. Brain 2013; 136:3461–3473
67.
Hodges JR, Mitchell J, Dawson K, et al: Semantic dementia: demography, familial factors and survival in a consecutive series of 100 cases. Brain 2010; 133:300–306
68.
Chan D, Anderson V, Pijnenburg Y, et al: The clinical profile of right temporal lobe atrophy. Brain 2009; 132:1287–1298
69.
Kamminga J, Kumfor F, Burrell JR, et al: Differentiating between right-lateralised semantic dementia and behavioural-variant frontotemporal dementia: an examination of clinical characteristics and emotion processing. J Neurol Neurosurg Psychiatry 2015; 86:1082–1088
70.
Coon EA, Whitwell JL, Parisi JE, et al: Right temporal variant frontotemporal dementia with motor neuron disease. J Clin Neurosci 2012; 19:85–91
71.
Edwards-Lee T, Miller BL, Benson DF, et al: The temporal variant of frontotemporal dementia. Brain 1997; 120:1027–1040
72.
Rosen HJ, Kramer JH, Gorno-Tempini ML, et al: Patterns of cerebral atrophy in primary progressive aphasia. Am J Geriatr Psychiatry 2002; 10:89–97
73.
Gorno-Tempini ML, Dronkers NF, Rankin KP, et al: Cognition and anatomy in three variants of primary progressive aphasia. Ann Neurol 2004; 55:335–346
74.
Mesulam MM: Primary progressive aphasia–a language-based dementia. N Engl J Med 2003; 349:1535–1542
75.
Lomen-Hoerth C: Clinical phenomenology and neuroimaging correlates in ALS-FTD. J Mol Neurosci 2011; 45:656–662
76.
Lillo P, Garcin B, Hornberger M, et al: Neurobehavioral features in frontotemporal dementia with amyotrophic lateral sclerosis. Arch Neurol 2010; 67:826–830
77.
Lomen-Hoerth C, Anderson T, Miller B: The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology 2002; 59:1077–1079
78.
Portet F, Cadilhac C, Touchon J, et al: Cognitive impairment in motor neuron disease with bulbar onset. Amyotroph Lateral Scler Other Motor Neuron Disord 2001; 2:23–29
79.
Hodges JR, Davies R, Xuereb J, et al: Survival in frontotemporal dementia. Neurology 2003; 61:349–354
80.
Armstrong MJ, Litvan I, Lang AE, et al: Criteria for the diagnosis of corticobasal degeneration. Neurology 2013; 80:496–503
81.
Lee SE, Rabinovici GD, Mayo MC, et al: Clinicopathological correlations in corticobasal degeneration. Ann Neurol 2011; 70:327–340
82.
Litvan I, Agid Y, Calne D, et al: Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology 1996; 47:1–9
83.
Dubois B, Slachevsky A, Pillon B, et al: “Applause sign” helps to discriminate PSP from FTD and PD. Neurology 2005; 64:2132–2133
84.
Williams DR, Lees AJ: Progressive supranuclear palsy: clinicopathological concepts and diagnostic challenges. Lancet Neurol 2009; 8:270–279
85.
Kanazawa M, Shimohata T, Toyoshima Y, et al: Cerebellar involvement in progressive supranuclear palsy: A clinicopathological study. Mov Disord 2009; 24:1312–1318
86.
Kim YE, Kang SY, Ma HI, et al: A visual rating scale for the hummingbird sign with adjustable diagnostic validity. J Parkinsons Dis 2015; 5:605–612
87.
Pearce JM: Pick’s disease. J Neurol Neurosurg Psychiatry 2003; 74:169
88.
Alzheimer A: Über eigenartige krankheitsfälle des späteren alters. Ztschr ges Neurol Psychiat 1911; 4:356–385.
89.
Neumann M, Sampathu DM, Kwong LK, et al: Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006; 314:130–133
90.
Sieben A, Van Langenhove T, Engelborghs S, et al: The genetics and neuropathology of frontotemporal lobar degeneration. Acta Neuropathol 2012; 124:353–372
91.
Sato-Harada R, Okabe S, Umeyama T, et al: Microtubule-associated proteins regulate microtubule function as the track for intracellular membrane organelle transports. Cell Struct Funct 1996; 21:283–295
92.
Buée L, Bussière T, Buée-Scherrer V, et al: Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev 2000; 33:95–130
93.
Irwin DJ, Brettschneider J, McMillan CT, et al: Deep clinical and neuropathological phenotyping of Pick disease. Ann Neurol 2016; 79:272–287
94.
Grossman M: Primary progressive aphasia: clinicopathological correlations. Nat Rev Neurol 2010; 6:88–97
95.
Dickson DW: Neuropathologic differentiation of progressive supranuclear palsy and corticobasal degeneration. J Neurol 1999; 246(Suppl 2):II6–II15
96.
Hauw JJ, Daniel SE, Dickson D, et al: Preliminary NINDS neuropathologic criteria for Steele-Richardson-Olszewski syndrome (progressive supranuclear palsy). Neurology 1994; 44:2015–2019
97.
Ahmed Z, Doherty KM, Silveira-Moriyama L, et al: Globular glial tauopathies (GGT) presenting with motor neuron disease or frontotemporal dementia: an emerging group of 4-repeat tauopathies. Acta Neuropathol 2011; 122:415–428
98.
Ahmed Z, Bigio EH, Budka H, et al: Globular glial tauopathies (GGT): consensus recommendations. Acta Neuropathol 2013; 126:537–544
99.
Braak H, Braak E: Cortical and subcortical argyrophilic grains characterize a disease associated with adult onset dementia. Neuropathol Appl Neurobiol 1989; 15:13–26
100.
Geser F, Martinez-Lage M, Robinson J, et al: Clinical and pathological continuum of multisystem TDP-43 proteinopathies. Arch Neurol 2009; 66:180–189
101.
Tan RH, Shepherd CE, Kril JJ, et al: Classification of FTLD-TDP cases into pathological subtypes using antibodies against phosphorylated and non-phosphorylated TDP43. Acta Neuropathol Commun 2013; 1:33
102.
Mackenzie IR, Neumann M, Baborie A, et al: A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol 2011; 122:111–113
103.
Harris JM, Gall C, Thompson JC, et al: Classification and pathology of primary progressive aphasia. Neurology 2013; 81:1832–1839
104.
Neumann M, Mackenzie IR, Cairns NJ, et al: TDP-43 in the ubiquitin pathology of frontotemporal dementia with VCP gene mutations. J Neuropathol Exp Neurol 2007; 66:152–157
105.
Lagier-Tourenne C, Cleveland DW: Rethinking ALS: the FUS about TDP-43. Cell 2009; 136:1001–1004
106.
Munoz DG, Neumann M, Kusaka H, et al: FUS pathology in basophilic inclusion body disease. Acta Neuropathol 2009; 118:617–627
107.
Neumann M, Roeber S, Kretzschmar HA, et al: Abundant FUS-immunoreactive pathology in neuronal intermediate filament inclusion disease. Acta Neuropathol 2009; 118:605–616
108.
Snowden JS, Hu Q, Rollinson S, et al: The most common type of FTLD-FUS (aFTLD-U) is associated with a distinct clinical form of frontotemporal dementia but is not related to mutations in the FUS gene. Acta Neuropathol 2011; 122:99–110
109.
Braak H, Braak E: Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991; 82:239–259
110.
DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al: Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011; 72:245–256
111.
Freibaum BD, Lu Y, Lopez-Gonzalez R, et al: GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 2015; 525:129–133
112.
Mori K, Weng SM, Arzberger T, et al: The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 2013; 339:1335–1338
113.
Cooper-Knock J, Kirby J, Highley R, et al: The Spectrum of C9orf72-mediated Neurodegeneration and Amyotrophic Lateral Sclerosis. Neurotherapeutics 2015; 12:326–339
114.
Whitwell JL, Weigand SD, Boeve BF, et al: Neuroimaging signatures of frontotemporal dementia genetics: C9ORF72, tau, progranulin and sporadics. Brain 2012; 135:794–806
115.
Sha SJ, Takada LT, Rankin KP, et al: Frontotemporal dementia due to C9ORF72 mutations: clinical and imaging features. Neurology 2012; 79:1002–1011
116.
Hutton M, Lendon CL, Rizzu P, et al: Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 1998; 393:702–705
117.
Janssen JC, Warrington EK, Morris HR, et al: Clinical features of frontotemporal dementia due to the intronic tau 10(+16) mutation. Neurology 2002; 58:1161–1168
118.
Petkau TL, Leavitt BR: Progranulin in neurodegenerative disease. Trends Neurosci 2014; 37:388–398
119.
Smith KR, Damiano J, Franceschetti S, et al: Strikingly different clinicopathological phenotypes determined by progranulin-mutation dosage. Am J Hum Genet 2012; 90:1102–1107
120.
Caroppo P, Le Ber I, Camuzat A, et al: Extensive white matter involvement in patients with frontotemporal lobar degeneration: think progranulin. JAMA Neurol 2014; 71:1562–1566
121.
Chen-Plotkin AS, Martinez-Lage M, Sleiman PM, et al: Genetic and clinical features of progranulin-associated frontotemporal lobar degeneration. Arch Neurol 2011; 68:488–497
122.
Cox LE, Ferraiuolo L, Goodall EF, et al: Mutations in CHMP2B in lower motor neuron predominant amyotrophic lateral sclerosis (ALS). PLoS One 2010; 5:e9872
123.
Urwin H, Authier A, Nielsen JE, et al: Disruption of endocytic trafficking in frontotemporal dementia with CHMP2B mutations. Hum Mol Genet 2010; 19:2228–2238
124.
Freischmidt A, Wieland T, Richter B, et al: Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat Neurosci 2015; 18:631–636
125.
Yan J, Deng HX, Siddique N, et al: Frameshift and novel mutations in FUS in familial amyotrophic lateral sclerosis and ALS/dementia. Neurology 2010; 75:807–814
126.
Gitcho MA, Bigio EH, Mishra M, et al: TARDBP 3′-UTR variant in autopsy-confirmed frontotemporal lobar degeneration with TDP-43 proteinopathy. Acta Neuropathol 2009; 118:633–645
127.
Iverson DJ, Gronseth GS, Reger MA, et al: Practice parameter update: evaluation and management of driving risk in dementia: report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2010; 74:1316–1324
128.
Teri L, Gibbons LE, McCurry SM, et al: Exercise plus behavioral management in patients with Alzheimer disease: a randomized controlled trial. JAMA 2003; 290:2015–2022
129.
Pitkälä KH, Pöysti MM, Laakkonen ML, et al: Effects of the Finnish Alzheimer disease exercise trial (FINALEX): a randomized controlled trial. JAMA Intern Med 2013; 173:894–901
130.
Sparks DL, Markesbery WR: Altered serotonergic and cholinergic synaptic markers in Pick’s disease. Arch Neurol 1991; 48:796–799
131.
Francis PT, Holmes C, Webster MT, et al: Preliminary neurochemical findings in non-Alzheimer dementia due to lobar atrophy. Dementia 1993; 4:172–177
132.
Swartz JR, Miller BL, Lesser IM, et al: Frontotemporal dementia: treatment response to serotonin selective reuptake inhibitors. J Clin Psychiatry 1997; 58:212–216
133.
Anneser JM, Jox RJ, Borasio GD: Inappropriate sexual behaviour in a case of ALS and FTD: successful treatment with sertraline. Amyotroph Lateral Scler 2007; 8:189–190
134.
Deakin JB, Rahman S, Nestor PJ, et al: Paroxetine does not improve symptoms and impairs cognition in frontotemporal dementia: a double-blind randomized controlled trial. Psychopharmacology (Berl) 2004; 172:400–408
135.
Herrmann N, Black SE, Chow T, et al: Serotonergic function and treatment of behavioral and psychological symptoms of frontotemporal dementia. Am J Geriatr Psychiatry 2012; 20:789–797
136.
Moretti R, Torre P, Antonello RM, et al: Frontotemporal dementia: paroxetine as a possible treatment of behavior symptoms. A randomized, controlled, open 14-month study. Eur Neurol 2003; 49:13–19
137.
Lebert F, Stekke W, Hasenbroekx C, et al: Frontotemporal dementia: a randomised, controlled trial with trazodone. Dement Geriatr Cogn Disord 2004; 17:355–359
138.
Rinne JO, Laine M, Kaasinen V, et al: Striatal dopamine transporter and extrapyramidal symptoms in frontotemporal dementia. Neurology 2002; 58:1489–1493
139.
Rahman S, Robbins TW, Hodges JR, et al: Methylphenidate (‘Ritalin’) can ameliorate abnormal risk-taking behavior in the frontal variant of frontotemporal dementia. Neuropsychopharmacology 2006; 31:651–658
140.
Huey ED, Garcia C, Wassermann EM, et al: Stimulant treatment of frontotemporal dementia in 8 patients. J Clin Psychiatry 2008; 69:1981–1982
141.
Jesso S, Morlog D, Ross S, et al: The effects of oxytocin on social cognition and behaviour in frontotemporal dementia. Brain 2011; 134:2493–2501
142.
Procter AW, Qurne M, Francis PT: Neurochemical features of frontotemporal dementia. Dement Geriatr Cogn Disord 1999; 10(Suppl 1):80–84
143.
Kertesz A, Morlog D, Light M, et al: Galantamine in frontotemporal dementia and primary progressive aphasia. Dement Geriatr Cogn Disord 2008; 25:178–185
144.
Moretti R, Torre P, Antonello RM, et al: Rivastigmine in frontotemporal dementia: an open-label study. Drugs Aging 2004; 21:931–937
145.
Mendez MF, Shapira JS, McMurtray A, et al: Preliminary findings: behavioral worsening on donepezil in patients with frontotemporal dementia. Am J Geriatr Psychiatry 2007; 15:84–87
146.
Boxer AL, Knopman DS, Kaufer DI, et al: Memantine in patients with frontotemporal lobar degeneration: a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol 2013; 12:149–156
147.
Vercelletto M, Boutoleau-Bretonnière C, Volteau C, et al: Memantine in behavioral variant frontotemporal dementia: negative results. J Alzheimers Dis 2011; 23:749–759
148.
Sink KM, Holden KF, Yaffe K: Pharmacological treatment of neuropsychiatric symptoms of dementia: a review of the evidence. JAMA 2005; 293:596–608
149.
Chow TW, Mendez MF: Goals in symptomatic pharmacologic management of frontotemporal lobar degeneration. Am J Alzheimers Dis Other Demen 2002; 17:267–272
150.
Cruz M, Marinho V, Fontenelle LF, et al: Topiramate may modulate alcohol abuse but not other compulsive behaviors in frontotemporal dementia: case report. Cogn Behav Neurol 2008; 21:104–106
151.
Boxer AL, Gold M, Huey E, et al: Frontotemporal degeneration, the next therapeutic frontier: molecules and animal models for frontotemporal degeneration drug development. Alzheimers Dement 2013; 9:176–188
152.
Lagier-Tourenne C, Baughn M, Rigo F, et al: Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc Natl Acad Sci USA 2013; 110:E4530–E4539
153.
Southwell AL, Skotte NH, Bennett CF, et al: Antisense oligonucleotide therapeutics for inherited neurodegenerative diseases. Trends Mol Med 2012; 18:634–643
154.
Decker DA, Heilman KM: Steroid treatment of primary progressive aphasia. Arch Neurol 2008; 65:1533–1535
Information & Authors
Information
Published In


History
Published in print: Fall 2016
Published online: 13 October 2016
Keywords
Authors
Funding Information
Dr. Ljubenkov reports no financial relationships with commercial interests. Dr. Miller reports receiving support from a UCSF/Quest Diagnostics Dementia Pathway Collaboration research grant. He also reports receiving royalties from Cambridge University Press, Guilford Publications, Inc., and Neurocase.
Metrics & Citations
Metrics
Citations
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
For more information or tips please see 'Downloading to a citation manager' in the Help menu.
View Options
View options
PDF/EPUB
View PDF/EPUBFigures
Figure 1. T1-Weighted MRI of a 62-Year-Old Patient With Behavioral-Variant Frontotemporal Dementia (bvFTD)
A: Axial view at the level of the genu of the corpus callosum in radiological orientation, showing asymmetric, right greater than left, atrophy of the frontal lobe, including the insular cortex (A1), lateral prefrontal cortex (A2), and anterior cingulate along with adjacent medial prefrontal cortex (A3). B: Coronal view at the level of the rostral corpus callosum in radiological orientation, showing asymmetric atrophy of the orbitofrontal cortex (B1), insular cortex (B2), lateral prefrontal cortex (B3), and anterior cingulate along with adjacent medial prefrontal cortex (B4). C: Sagittal view of the mesial right hemisphere, showing frontal atrophy, including mesial prefrontal cortex (C1), pregenual cingulate (C2), orbitofrontal cortex (C3), and subgenual cingulate (C4).
Figure 2. Axial T1-Weighted MRI of a 63-Year-Old Patient with Semantic-Variant Primary Progressive Aphasia (svPPA) in Radiological Orientationa
aAsymmetric left greater than right anterior temporal lobe atrophy noted by an arrow
Figure 3. T1-Weighted MRI of a 70-Year-Old Patient With Nonfluent/Agrammatic-Variant Primary Progressive Aphasia (nfvPPA)
A: Axial view in radiological orientation showing asymmetric, left greater than right atrophy of the frontal operculum (arrow). B: Coronal view showing left greater than right atrophy of the anterior perisylvian region (arrow). C: Sagittal view of the left lateral cerebral hemisphere, showing left anterior perisylvian atrophy, including the frontal operculum (arrow).
Figure 4. Sagittal T1-Weighted MRI of a 71-Year-Old Patient With Progressive Supranuclear Palsy (PSP)a
aImage shows “hummingbird sign,” including an atrophic midbrain tegmentum (dashed arrow) resembling a hummingbird’s head and a relatively preserved basis pontis (solid arrow) that resembles the hummingbird’s belly.
Tables
Media
References
References
1.
Brun A, Liu X, Erikson C: Synapse loss and gliosis in the molecular layer of the cerebral cortex in Alzheimer’s disease and in frontal lobe degeneration. Neurodegeneration 1995; 4:171–177
2.
Mercy L, Hodges JR, Dawson K, et al: Incidence of early-onset dementias in Cambridgeshire, United Kingdom. Neurology 2008; 71:1496–1499
3.
Ratnavalli E, Brayne C, Dawson K, et al: The prevalence of frontotemporal dementia. Neurology 2002; 58:1615–1621
4.
Johnson JK, Diehl J, Mendez MF, et al: Frontotemporal lobar degeneration: demographic characteristics of 353 patients. Arch Neurol 2005; 62:925–930
5.
Knopman DS, Roberts RO: Estimating the number of persons with frontotemporal lobar degeneration in the US population. J Mol Neurosci 2011; 45:330–335
6.
Coyle-Gilchrist ITS, Dick KM, Patterson K, et al: Prevalence, characteristics, and survival of frontotemporal lobar degeneration syndromes. Neurology 2016; 86:1736–1743
7.
Onyike CU, Diehl-Schmid J. The epidemiology of frontotemporal dementia. Int Rev Psychiatry 2013; 25:130–137.
8.
Rosso SM, Donker Kaat L, Baks T, et al: Frontotemporal dementia in the Netherlands: patient characteristics and prevalence estimates from a population-based study. Brain 2003; 126:2016–2022
9.
Ikejima C, Yasuno F, Mizukami K, et al: Prevalence and causes of early-onset dementia in Japan: a population-based study. Stroke 2009; 40:2709–2714
10.
Borroni B, Alberici A, Grassi M, et al: Is frontotemporal lobar degeneration a rare disorder? Evidence from a preliminary study in Brescia County, Italy. J Alzheimers Dis 2010; 19:111–116
11.
Goldman JS, Farmer JM, Wood EM, et al: Comparison of family histories in FTLD subtypes and related tauopathies. Neurology 2005; 65:1817–1819
12.
Le Ber I, Guedj E, Gabelle A, et al:Demographic, neurological and behavioural characteristics and brain perfusion SPECT in frontal variant of frontotemporal dementia. Brain 2006; 129:3051–3065
13.
Roberson ED, Hesse JH, Rose KD, et al: Frontotemporal dementia progresses to death faster than Alzheimer disease. Neurology 2005; 65:719–725
14.
Kertesz A, McMonagle P, Blair M, et al: The evolution and pathology of frontotemporal dementia. Brain 2005; 128:1996–2005
15.
Woollacott IO, Rohrer JD: The clinical spectrum of sporadic and familial forms of frontotemporal dementia. J Neurochem (Epub May 3, 2016)
16.
Schroeter ML, Raczka K, Neumann J, et al: Neural networks in frontotemporal dementia—a meta-analysis. Neurobiol Aging 2008; 29:418–426
17.
Seeley WW, Crawford RK, Zhou J, et al: Neurodegenerative diseases target large-scale human brain networks. Neuron 2009; 62:42–52
18.
Rademakers R, Neumann M, Mackenzie IR: Advances in understanding the molecular basis of frontotemporal dementia. Nat Rev Neurol 2012; 8:423–434
19.
Rascovsky K, Hodges JR, Knopman D, et al: Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 2011; 134:2456–2477
20.
Rosen HJ, Allison SC, Schauer GF, et al: Neuroanatomical correlates of behavioural disorders in dementia. Brain 2005; 128:2612–2625
21.
Seeley WW, Crawford R, Rascovsky K, et al: Frontal paralimbic network atrophy in very mild behavioral variant frontotemporal dementia. Arch Neurol 2008; 65:249–255
22.
Mendez MF, Lauterbach EC, Sampson SM: An evidence-based review of the psychopathology of frontotemporal dementia: a report of the ANPA Committee on Research. J Neuropsychiatry Clin Neurosci 2008; 20:130–149
23.
Massimo L, Powers C, Moore P, et al: Neuroanatomy of apathy and disinhibition in frontotemporal lobar degeneration. Dement Geriatr Cogn Disord 2009; 27:96–104
24.
Holroyd CB, Yeung N: Motivation of extended behaviors by anterior cingulate cortex. Trends Cogn Sci 2012; 16:122–128
25.
Sollberger M, Rosen HJ, Shany-Ur T, et al: Neural substrates of socioemotional self-awareness in neurodegenerative disease. Brain Behav 2014; 4:201–214
26.
Tranel D, Bechara A, Denburg NL: Asymmetric functional roles of right and left ventromedial prefrontal cortices in social conduct, decision-making, and emotional processing. Cortex 2002; 38:589–612
27.
Sturm VE, Sollberger M, Seeley WW, Rankin KP, Ascher EA, Rosen HJ, et al: Role of right pregenual anterior cingulate cortex in self-conscious emotional reactivity. Soc Cogn Affect Neurosci 2013; 8:468–474
28.
Eckart JA, Sturm VE, Miller BL, et al: Diminished disgust reactivity in behavioral variant frontotemporal dementia. Neuropsychologia 2012; 50:786–790
29.
Wright P, He G, Shapira NA, et al: Disgust and the insula: fMRI responses to pictures of mutilation and contamination. Neuroreport 2004; 15:2347–2351
30.
Ahmed RM, Kaizik C, Irish M, et al: Characterizing sexual behavior in frontotemporal dementia. J Alzheimers Dis 2015; 46:677–686
31.
Granadillo ED, Mendez MF: Pathological joking or Witzelsucht revisited. J Neuropsychiatry Clin Neurosci 2016; 28:162–167
32.
Williamson C, Alcantar O, Rothlind J, et al: Standardised measurement of self-awareness deficits in FTD and AD. J Neurol Neurosurg Psychiatry 2010; 81:140–145
33.
Seeley WW: Anterior insula degeneration in frontotemporal dementia. Brain Struct Funct 2010; 214:465–475
34.
Rankin KP, Gorno-Tempini ML, Allison SC, et al: Structural anatomy of empathy in neurodegenerative disease. Brain 2006; 129:2945–2956
35.
Perry DC, Whitwell JL, Boeve BF, et al: Voxel-based morphometry in patients with obsessive-compulsive behaviors in behavioral variant frontotemporal dementia. Eur J Neurol 2012; 19:911–917
36.
Woolley JD, Gorno-Tempini ML, Seeley WW, et al: Binge eating is associated with right orbitofrontal-insular-striatal atrophy in frontotemporal dementia. Neurology 2007; 69:1424–1433
37.
Piguet O: Eating disturbance in behavioural-variant frontotemporal dementia. J Mol Neurosci 2011; 45:589–593
38.
Henry ML, Wilson SM, Ogar JM, et al: Neuropsychological, behavioral, and anatomical evolution in right temporal variant frontotemporal dementia: a longitudinal and post-mortem single case analysis. Neurocase 2014; 20:100–109
39.
Kramer JH, Jurik J, Sha SJ, et al: Distinctive neuropsychological patterns in frontotemporal dementia, semantic dementia, and Alzheimer disease. Cogn Behav Neurol 2003; 16:211–218
40.
Huey ED, Goveia EN, Paviol S, et al: Executive dysfunction in frontotemporal dementia and corticobasal syndrome. Neurology 2009; 72:453–459
41.
Torralva T, Roca M, Gleichgerrcht E, et al: A neuropsychological battery to detect specific executive and social cognitive impairments in early frontotemporal dementia. Brain 2009; 132:1299–1309
42.
Hornberger M, Piguet O, Graham AJ, et al: How preserved is episodic memory in behavioral variant frontotemporal dementia? Neurology 2010; 74:472–479
43.
Rosen HJ, Narvaez JM, Hallam B, et al: Neuropsychological and functional measures of severity in Alzheimer disease, frontotemporal dementia, and semantic dementia. Alzheimer Dis Assoc Disord 2004; 18:202–207
44.
Perry RJ, Graham A, Williams G, et al: Patterns of frontal lobe atrophy in frontotemporal dementia: a volumetric MRI study. Dement Geriatr Cogn Disord 2006; 22:278–287
45.
Rosen HJ, Gorno-Tempini ML, Goldman WP, et al: Patterns of brain atrophy in frontotemporal dementia and semantic dementia. Neurology 2002; 58:198–208
46.
Seeley WW: Selective functional, regional, and neuronal vulnerability in frontotemporal dementia. Curr Opin Neurol 2008; 21:701–707
47.
Mendez MF, Shapira JS, McMurtray A, et al: Accuracy of the clinical evaluation for frontotemporal dementia. Arch Neurol 2007; 64:830–835
48.
Foster NL, Heidebrink JL, Clark CM, et al: FDG-PET improves accuracy in distinguishing frontotemporal dementia and Alzheimer’s disease. Brain 2007; 130:2616–2635
49.
Ossenkoppele R, Pijnenburg YA, Perry DC, et al: The behavioural/dysexecutive variant of Alzheimer’s disease: clinical, neuroimaging and pathological features. Brain 2015; 138:2732–2749
50.
Ewers M, Mattsson N, Minthon L, et al: CSF biomarkers for the differential diagnosis of Alzheimer’s disease: A large-scale international multicenter study. Alzheimers Dement 2015; 11:1306–1315
51.
McKhann GM, Knopman DS, Chertkow H, et al: The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 2011; 7:263–269
52.
Ossenkoppele R, Jansen WJ, Rabinovici GD, et al: Prevalence of amyloid PET positivity in dementia syndromes: a meta-analysis. JAMA 2015; 313:1939–1949
53.
Mendez MF, Shapira JS, Woods RJ, et al: Psychotic symptoms in frontotemporal dementia: prevalence and review. Dement Geriatr Cogn Disord 2008; 25:206–211
54.
Berg D, Postuma RB, Adler CH, et al: MDS research criteria for prodromal Parkinson’s disease. Mov Disord 2015; 30:1600–1611
55.
Rascovsky K, Grossman M: Clinical diagnostic criteria and classification controversies in frontotemporal lobar degeneration. Int Rev Psychiatry 2013; 25:145–158
56.
Khan BK, Yokoyama JS, Takada LT, et al: Atypical, slowly progressive behavioural variant frontotemporal dementia associated with C9ORF72 hexanucleotide expansion. J Neurol Neurosurg Psychiatry 2012; 83:358–364
57.
Velakoulis D, Walterfang M, Mocellin R, et al: Frontotemporal dementia presenting as schizophrenia-like psychosis in young people: clinicopathological series and review of cases. Br J Psychiatry 2009; 194:298–305
58.
Snowden JS, Rollinson S, Thompson JC, et al: Distinct clinical and pathological characteristics of frontotemporal dementia associated with C9ORF72 mutations. Brain 2012; 135:693–708
59.
Lanata SC, Miller BL: The behavioural variant frontotemporal dementia (bvFTD) syndrome in psychiatry. J Neurol Neurosurg Psychiatry 2016; 87:501–511
60.
Galimberti D, Dell’Osso B, Fenoglio C, et al: Progranulin gene variability and plasma levels in bipolar disorder and schizophrenia. PLoS One 2012; 7:e32164
61.
Snowden JS, Adams J, Harris J, et al: Distinct clinical and pathological phenotypes in frontotemporal dementia associated with MAPT, PGRN and C9orf72 mutations. Amyotroph Lateral Scler Frontotemporal Degener 2015; 16:497–505
62.
Gorno-Tempini ML, Hillis AE, Weintraub S, et al: Classification of primary progressive aphasia and its variants. Neurology 2011; 76:1006–1014
63.
Mesulam M, Wicklund A, Johnson N, et al: Alzheimer and frontotemporal pathology in subsets of primary progressive aphasia. Ann Neurol 2008; 63:709–719
64.
Guo CC, Gorno-Tempini ML, Gesierich B, et al: Anterior temporal lobe degeneration produces widespread network-driven dysfunction. Brain 2013; 136:2979–2991
65.
Seeley WW, Bauer AM, Miller BL, et al: The natural history of temporal variant frontotemporal dementia. Neurology 2005; 64:1384–1390
66.
Miller ZA, Mandelli ML, Rankin KP, et al: Handedness and language learning disability differentially distribute in progressive aphasia variants. Brain 2013; 136:3461–3473
67.
Hodges JR, Mitchell J, Dawson K, et al: Semantic dementia: demography, familial factors and survival in a consecutive series of 100 cases. Brain 2010; 133:300–306
68.
Chan D, Anderson V, Pijnenburg Y, et al: The clinical profile of right temporal lobe atrophy. Brain 2009; 132:1287–1298
69.
Kamminga J, Kumfor F, Burrell JR, et al: Differentiating between right-lateralised semantic dementia and behavioural-variant frontotemporal dementia: an examination of clinical characteristics and emotion processing. J Neurol Neurosurg Psychiatry 2015; 86:1082–1088
70.
Coon EA, Whitwell JL, Parisi JE, et al: Right temporal variant frontotemporal dementia with motor neuron disease. J Clin Neurosci 2012; 19:85–91
71.
Edwards-Lee T, Miller BL, Benson DF, et al: The temporal variant of frontotemporal dementia. Brain 1997; 120:1027–1040
72.
Rosen HJ, Kramer JH, Gorno-Tempini ML, et al: Patterns of cerebral atrophy in primary progressive aphasia. Am J Geriatr Psychiatry 2002; 10:89–97
73.
Gorno-Tempini ML, Dronkers NF, Rankin KP, et al: Cognition and anatomy in three variants of primary progressive aphasia. Ann Neurol 2004; 55:335–346
74.
Mesulam MM: Primary progressive aphasia–a language-based dementia. N Engl J Med 2003; 349:1535–1542
75.
Lomen-Hoerth C: Clinical phenomenology and neuroimaging correlates in ALS-FTD. J Mol Neurosci 2011; 45:656–662
76.
Lillo P, Garcin B, Hornberger M, et al: Neurobehavioral features in frontotemporal dementia with amyotrophic lateral sclerosis. Arch Neurol 2010; 67:826–830
77.
Lomen-Hoerth C, Anderson T, Miller B: The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology 2002; 59:1077–1079
78.
Portet F, Cadilhac C, Touchon J, et al: Cognitive impairment in motor neuron disease with bulbar onset. Amyotroph Lateral Scler Other Motor Neuron Disord 2001; 2:23–29
79.
Hodges JR, Davies R, Xuereb J, et al: Survival in frontotemporal dementia. Neurology 2003; 61:349–354
80.
Armstrong MJ, Litvan I, Lang AE, et al: Criteria for the diagnosis of corticobasal degeneration. Neurology 2013; 80:496–503
81.
Lee SE, Rabinovici GD, Mayo MC, et al: Clinicopathological correlations in corticobasal degeneration. Ann Neurol 2011; 70:327–340
82.
Litvan I, Agid Y, Calne D, et al: Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology 1996; 47:1–9
83.
Dubois B, Slachevsky A, Pillon B, et al: “Applause sign” helps to discriminate PSP from FTD and PD. Neurology 2005; 64:2132–2133
84.
Williams DR, Lees AJ: Progressive supranuclear palsy: clinicopathological concepts and diagnostic challenges. Lancet Neurol 2009; 8:270–279
85.
Kanazawa M, Shimohata T, Toyoshima Y, et al: Cerebellar involvement in progressive supranuclear palsy: A clinicopathological study. Mov Disord 2009; 24:1312–1318
86.
Kim YE, Kang SY, Ma HI, et al: A visual rating scale for the hummingbird sign with adjustable diagnostic validity. J Parkinsons Dis 2015; 5:605–612
87.
Pearce JM: Pick’s disease. J Neurol Neurosurg Psychiatry 2003; 74:169
88.
Alzheimer A: Über eigenartige krankheitsfälle des späteren alters. Ztschr ges Neurol Psychiat 1911; 4:356–385.
89.
Neumann M, Sampathu DM, Kwong LK, et al: Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006; 314:130–133
90.
Sieben A, Van Langenhove T, Engelborghs S, et al: The genetics and neuropathology of frontotemporal lobar degeneration. Acta Neuropathol 2012; 124:353–372
91.
Sato-Harada R, Okabe S, Umeyama T, et al: Microtubule-associated proteins regulate microtubule function as the track for intracellular membrane organelle transports. Cell Struct Funct 1996; 21:283–295
92.
Buée L, Bussière T, Buée-Scherrer V, et al: Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev 2000; 33:95–130
93.
Irwin DJ, Brettschneider J, McMillan CT, et al: Deep clinical and neuropathological phenotyping of Pick disease. Ann Neurol 2016; 79:272–287
94.
Grossman M: Primary progressive aphasia: clinicopathological correlations. Nat Rev Neurol 2010; 6:88–97
95.
Dickson DW: Neuropathologic differentiation of progressive supranuclear palsy and corticobasal degeneration. J Neurol 1999; 246(Suppl 2):II6–II15
96.
Hauw JJ, Daniel SE, Dickson D, et al: Preliminary NINDS neuropathologic criteria for Steele-Richardson-Olszewski syndrome (progressive supranuclear palsy). Neurology 1994; 44:2015–2019
97.
Ahmed Z, Doherty KM, Silveira-Moriyama L, et al: Globular glial tauopathies (GGT) presenting with motor neuron disease or frontotemporal dementia: an emerging group of 4-repeat tauopathies. Acta Neuropathol 2011; 122:415–428
98.
Ahmed Z, Bigio EH, Budka H, et al: Globular glial tauopathies (GGT): consensus recommendations. Acta Neuropathol 2013; 126:537–544
99.
Braak H, Braak E: Cortical and subcortical argyrophilic grains characterize a disease associated with adult onset dementia. Neuropathol Appl Neurobiol 1989; 15:13–26
100.
Geser F, Martinez-Lage M, Robinson J, et al: Clinical and pathological continuum of multisystem TDP-43 proteinopathies. Arch Neurol 2009; 66:180–189
101.
Tan RH, Shepherd CE, Kril JJ, et al: Classification of FTLD-TDP cases into pathological subtypes using antibodies against phosphorylated and non-phosphorylated TDP43. Acta Neuropathol Commun 2013; 1:33
102.
Mackenzie IR, Neumann M, Baborie A, et al: A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol 2011; 122:111–113
103.
Harris JM, Gall C, Thompson JC, et al: Classification and pathology of primary progressive aphasia. Neurology 2013; 81:1832–1839
104.
Neumann M, Mackenzie IR, Cairns NJ, et al: TDP-43 in the ubiquitin pathology of frontotemporal dementia with VCP gene mutations. J Neuropathol Exp Neurol 2007; 66:152–157
105.
Lagier-Tourenne C, Cleveland DW: Rethinking ALS: the FUS about TDP-43. Cell 2009; 136:1001–1004
106.
Munoz DG, Neumann M, Kusaka H, et al: FUS pathology in basophilic inclusion body disease. Acta Neuropathol 2009; 118:617–627
107.
Neumann M, Roeber S, Kretzschmar HA, et al: Abundant FUS-immunoreactive pathology in neuronal intermediate filament inclusion disease. Acta Neuropathol 2009; 118:605–616
108.
Snowden JS, Hu Q, Rollinson S, et al: The most common type of FTLD-FUS (aFTLD-U) is associated with a distinct clinical form of frontotemporal dementia but is not related to mutations in the FUS gene. Acta Neuropathol 2011; 122:99–110
109.
Braak H, Braak E: Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991; 82:239–259
110.
DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al: Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011; 72:245–256
111.
Freibaum BD, Lu Y, Lopez-Gonzalez R, et al: GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 2015; 525:129–133
112.
Mori K, Weng SM, Arzberger T, et al: The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 2013; 339:1335–1338
113.
Cooper-Knock J, Kirby J, Highley R, et al: The Spectrum of C9orf72-mediated Neurodegeneration and Amyotrophic Lateral Sclerosis. Neurotherapeutics 2015; 12:326–339
114.
Whitwell JL, Weigand SD, Boeve BF, et al: Neuroimaging signatures of frontotemporal dementia genetics: C9ORF72, tau, progranulin and sporadics. Brain 2012; 135:794–806
115.
Sha SJ, Takada LT, Rankin KP, et al: Frontotemporal dementia due to C9ORF72 mutations: clinical and imaging features. Neurology 2012; 79:1002–1011
116.
Hutton M, Lendon CL, Rizzu P, et al: Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 1998; 393:702–705
117.
Janssen JC, Warrington EK, Morris HR, et al: Clinical features of frontotemporal dementia due to the intronic tau 10(+16) mutation. Neurology 2002; 58:1161–1168
118.
Petkau TL, Leavitt BR: Progranulin in neurodegenerative disease. Trends Neurosci 2014; 37:388–398
119.
Smith KR, Damiano J, Franceschetti S, et al: Strikingly different clinicopathological phenotypes determined by progranulin-mutation dosage. Am J Hum Genet 2012; 90:1102–1107
120.
Caroppo P, Le Ber I, Camuzat A, et al: Extensive white matter involvement in patients with frontotemporal lobar degeneration: think progranulin. JAMA Neurol 2014; 71:1562–1566
121.
Chen-Plotkin AS, Martinez-Lage M, Sleiman PM, et al: Genetic and clinical features of progranulin-associated frontotemporal lobar degeneration. Arch Neurol 2011; 68:488–497
122.
Cox LE, Ferraiuolo L, Goodall EF, et al: Mutations in CHMP2B in lower motor neuron predominant amyotrophic lateral sclerosis (ALS). PLoS One 2010; 5:e9872
123.
Urwin H, Authier A, Nielsen JE, et al: Disruption of endocytic trafficking in frontotemporal dementia with CHMP2B mutations. Hum Mol Genet 2010; 19:2228–2238
124.
Freischmidt A, Wieland T, Richter B, et al: Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat Neurosci 2015; 18:631–636
125.
Yan J, Deng HX, Siddique N, et al: Frameshift and novel mutations in FUS in familial amyotrophic lateral sclerosis and ALS/dementia. Neurology 2010; 75:807–814
126.
Gitcho MA, Bigio EH, Mishra M, et al: TARDBP 3′-UTR variant in autopsy-confirmed frontotemporal lobar degeneration with TDP-43 proteinopathy. Acta Neuropathol 2009; 118:633–645
127.
Iverson DJ, Gronseth GS, Reger MA, et al: Practice parameter update: evaluation and management of driving risk in dementia: report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2010; 74:1316–1324
128.
Teri L, Gibbons LE, McCurry SM, et al: Exercise plus behavioral management in patients with Alzheimer disease: a randomized controlled trial. JAMA 2003; 290:2015–2022
129.
Pitkälä KH, Pöysti MM, Laakkonen ML, et al: Effects of the Finnish Alzheimer disease exercise trial (FINALEX): a randomized controlled trial. JAMA Intern Med 2013; 173:894–901
130.
Sparks DL, Markesbery WR: Altered serotonergic and cholinergic synaptic markers in Pick’s disease. Arch Neurol 1991; 48:796–799
131.
Francis PT, Holmes C, Webster MT, et al: Preliminary neurochemical findings in non-Alzheimer dementia due to lobar atrophy. Dementia 1993; 4:172–177
132.
Swartz JR, Miller BL, Lesser IM, et al: Frontotemporal dementia: treatment response to serotonin selective reuptake inhibitors. J Clin Psychiatry 1997; 58:212–216
133.
Anneser JM, Jox RJ, Borasio GD: Inappropriate sexual behaviour in a case of ALS and FTD: successful treatment with sertraline. Amyotroph Lateral Scler 2007; 8:189–190
134.
Deakin JB, Rahman S, Nestor PJ, et al: Paroxetine does not improve symptoms and impairs cognition in frontotemporal dementia: a double-blind randomized controlled trial. Psychopharmacology (Berl) 2004; 172:400–408
135.
Herrmann N, Black SE, Chow T, et al: Serotonergic function and treatment of behavioral and psychological symptoms of frontotemporal dementia. Am J Geriatr Psychiatry 2012; 20:789–797
136.
Moretti R, Torre P, Antonello RM, et al: Frontotemporal dementia: paroxetine as a possible treatment of behavior symptoms. A randomized, controlled, open 14-month study. Eur Neurol 2003; 49:13–19
137.
Lebert F, Stekke W, Hasenbroekx C, et al: Frontotemporal dementia: a randomised, controlled trial with trazodone. Dement Geriatr Cogn Disord 2004; 17:355–359
138.
Rinne JO, Laine M, Kaasinen V, et al: Striatal dopamine transporter and extrapyramidal symptoms in frontotemporal dementia. Neurology 2002; 58:1489–1493
139.
Rahman S, Robbins TW, Hodges JR, et al: Methylphenidate (‘Ritalin’) can ameliorate abnormal risk-taking behavior in the frontal variant of frontotemporal dementia. Neuropsychopharmacology 2006; 31:651–658
140.
Huey ED, Garcia C, Wassermann EM, et al: Stimulant treatment of frontotemporal dementia in 8 patients. J Clin Psychiatry 2008; 69:1981–1982
141.
Jesso S, Morlog D, Ross S, et al: The effects of oxytocin on social cognition and behaviour in frontotemporal dementia. Brain 2011; 134:2493–2501
142.
Procter AW, Qurne M, Francis PT: Neurochemical features of frontotemporal dementia. Dement Geriatr Cogn Disord 1999; 10(Suppl 1):80–84
143.
Kertesz A, Morlog D, Light M, et al: Galantamine in frontotemporal dementia and primary progressive aphasia. Dement Geriatr Cogn Disord 2008; 25:178–185
144.
Moretti R, Torre P, Antonello RM, et al: Rivastigmine in frontotemporal dementia: an open-label study. Drugs Aging 2004; 21:931–937
145.
Mendez MF, Shapira JS, McMurtray A, et al: Preliminary findings: behavioral worsening on donepezil in patients with frontotemporal dementia. Am J Geriatr Psychiatry 2007; 15:84–87
146.
Boxer AL, Knopman DS, Kaufer DI, et al: Memantine in patients with frontotemporal lobar degeneration: a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol 2013; 12:149–156
147.
Vercelletto M, Boutoleau-Bretonnière C, Volteau C, et al: Memantine in behavioral variant frontotemporal dementia: negative results. J Alzheimers Dis 2011; 23:749–759
148.
Sink KM, Holden KF, Yaffe K: Pharmacological treatment of neuropsychiatric symptoms of dementia: a review of the evidence. JAMA 2005; 293:596–608
149.
Chow TW, Mendez MF: Goals in symptomatic pharmacologic management of frontotemporal lobar degeneration. Am J Alzheimers Dis Other Demen 2002; 17:267–272
150.
Cruz M, Marinho V, Fontenelle LF, et al: Topiramate may modulate alcohol abuse but not other compulsive behaviors in frontotemporal dementia: case report. Cogn Behav Neurol 2008; 21:104–106
151.
Boxer AL, Gold M, Huey E, et al: Frontotemporal degeneration, the next therapeutic frontier: molecules and animal models for frontotemporal degeneration drug development. Alzheimers Dement 2013; 9:176–188
152.
Lagier-Tourenne C, Baughn M, Rigo F, et al: Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc Natl Acad Sci USA 2013; 110:E4530–E4539
153.
Southwell AL, Skotte NH, Bennett CF, et al: Antisense oligonucleotide therapeutics for inherited neurodegenerative diseases. Trends Mol Med 2012; 18:634–643
154.
Decker DA, Heilman KM: Steroid treatment of primary progressive aphasia. Arch Neurol 2008; 65:1533–1535