Major depressive disorder is a significant pediatric health problem with a point prevalence ranging from 3.5% to 8% and a lifetime prevalence approaching 20% by the end of adolescence (
1,
2). Adolescent major depression is associated with considerable morbidity, including poor social and scholastic functioning, unplanned pregnancy, and increased risk of physical illness and substance abuse (
3,
4). It is also linked with significant mortality, with increased risk for suicide, which has recently moved from the third to the second leading cause of death in 15- to 24-year-olds (
5). Although currently available treatments for adolescent depression can be quite effective for many, there remains a substantial proportion of patients who fail to experience a sustained remission with the current treatment options (
6). Approximately 40% of patients remain depressed despite treatment with selective serotonin reuptake inhibitors (SSRIs) (
7), and while another half of this SSRI-resistant population can be expected to remit after switching medications and adding psychotherapy (
8), this leaves approximately 1 out of 5 patients who do not achieve depression relief with the currently available treatment strategies. Of additional concern is that 1 in 4 of those who do respond can be expected to relapse within a year (
9). These data highlight the need for novel therapies targeting distinct neurochemical systems that are thoughtfully considered in a developmental context.
The glutamate system is implicated in the pathophysiology of major depressive disorder (
10). Ketamine, a glutamatergic modulator, shows rapid antidepressant effects in adults, with an odds ratio of 9.87 for response 24 hours after treatment (
11). Ketamine improves a range of depressive symptoms in adults, and it notably reduces anhedonia (
12), a symptom associated with poor therapeutic response in adolescent major depression (
13). Ketamine also appears to have effectiveness in reducing suicidality in adults (
14), a dimension of adolescent major depression that has shown controversial associations with SSRIs (
15), making ketamine a potentially valuable new treatment option for the adolescent population. While the glutamate system continues to mature in adolescence (
16), preclinical data show that ketamine reverses depressive phenotypes in adolescent rats (
17).
Given its safety and success in treating adult major depression, ketamine is beginning to be considered for use in severe, treatment-refractory affective disorders in adolescence (
18). While ketamine has been used commonly as a pediatric anesthetic for many years (
19), there is little data on the safety, efficacy, or effectiveness of the psychiatric use of ketamine in children or adolescents. Case reports suggested early potential efficacy of intravenous ketamine in adolescent treatment-resistant depression (
20) and intranasal ketamine in bipolar depression (
18). Recently a small open-label trial of intravenous ketamine in 13 adolescents with treatment-resistant depression also suggested that ketamine might be effective in this population (
21), although the lack of placebo in the study complicates interpretation of the results. Placebo response rates are high in depression trials, and are particularly high in pediatric depression trials (
22), and it is critical to discriminate genuine drug effects from nonspecific effects. Thus, carefully monitored placebo-controlled prospective studies of ketamine’s safety and efficacy are needed to provide responsible, evidence-based care to children and adolescents.
Methods
Participants
Adolescents 13 to 17 years of age were recruited through physician referral or direct inquiries from families through the study’s ClinicalTrials.gov listing. Participants were enrolled at the Yale Child Study Center (New Haven, Conn.) between May 2016 and September 2018. To be eligible, participants had to have a primary DSM-5 diagnosis of major depressive disorder as determined by the Schedule for Affective Disorders and Schizophrenia for School-Age Children (K-SADS) and have a score >40 on the Children’s Depression Rating Scale–Revised (CDRS-R) (
23). Adolescents also must have failed to benefit from at least one prior 8-week trial of a standard antidepressant medication at a therapeutic dose in order to be considered treatment resistant.
Exclusion criteria were a lifetime history of a psychotic disorder or mania, autism spectrum disorder, intellectual disability, substance use disorder (excluding tobacco), or active suicidal or homicidal ideation on presentation requiring inpatient hospitalization. Participants were required to remain on stable dosing of their current psychiatric medication regimen for the 4 weeks prior to the first infusion and during the 4-week trial itself. All families were made aware of potential evidence-based and alternative treatments for adolescent depression, and all enrolled participants reported having previously received an antidepressant trial of adequate dosage and duration (
6). Individuals who did not respond to antidepressant treatment or had significant side effects to past antidepressant treatment were not required to be taking an antidepressant during the trial. All participants underwent a physical examination and laboratory screening, including routine chemistry and hematology tests, urine toxicology, and electrocardiography, and female participants received urine pregnancy testing.
The institutional review board at Yale School of Medicine approved the study. Written informed consent was obtained from the adolescents’ parents, and written informed assent from the adolescents, after all parties received a complete description of the study. Participants were compensated $150 for completing the study, and parents received parking vouchers. A subset of participants received an additional $100 to complete neuroimaging.
Procedures
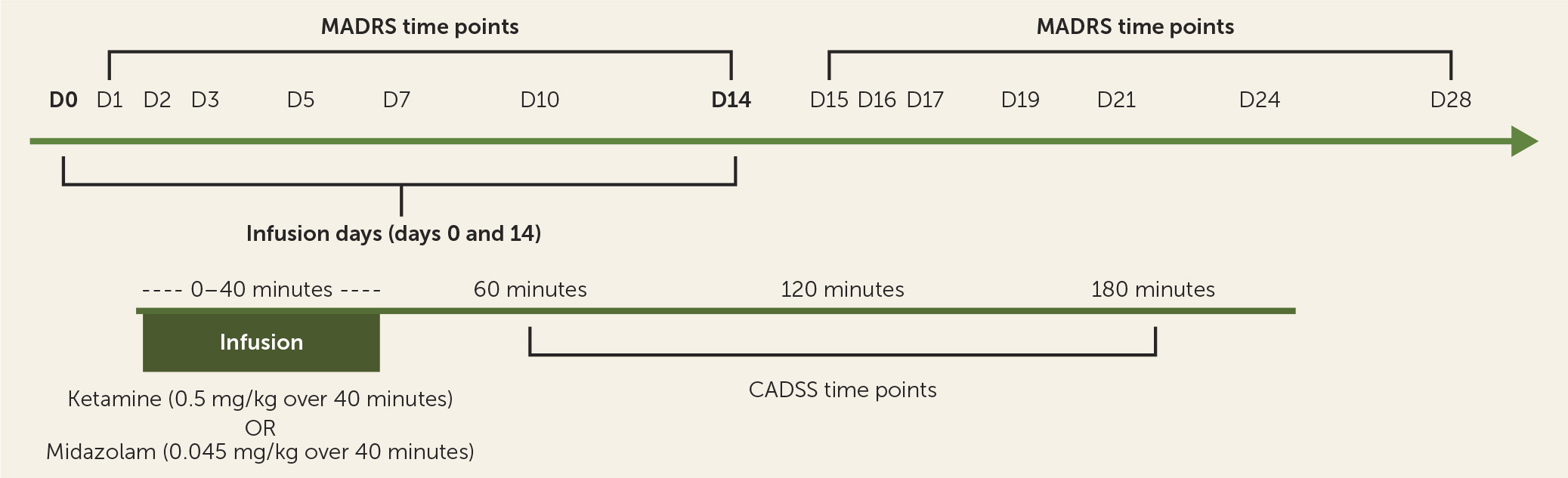
Participants fasted overnight and presented in the morning to Yale New Haven Hospital’s Hospital Research Unit. Two intravenous catheters were placed in each arm, one for medication infusion and the other for blood draws for pharmacokinetics. An ECG was performed, and pulse, blood pressure, and pulse oximetry were checked every 5 minutes during the infusion and every 15 minutes through 190 minutes after start of the infusion. A physician from the Yale Pediatric Sedation Service was present during all infusions and monitored the participant. A trained rater administered the Clinician-Administered Dissociative States Scale (CADSS) (
24) to assess side effects 1 and 2 hours after start of infusion. A different trained rater, absent during the infusion and blinded to the intrainfusion side effect ratings, administered rating scales at preinfusion baseline and eight postinfusion time points: at 3 hours, 1 day, 2 days, 3 days, 5 days, 7 days, 10 days, and 14 days.
The Yale Investigational Drug Service randomized the treatment order, with participants receiving a single infusion of either ketamine hydrochloride (0.5 mg/kg) or midazolam (0.045 mg/kg) on day 1, and the alternate compound 2 weeks later (
Figure 1). One participant received the second infusion 20 days after the first because of scheduling constraints, but otherwise there was no adjustment of timing between the first and second infusions. Midazolam was chosen as an active placebo, in keeping with its similar pharmacokinetic profile and precedent (
25) as a reasonable comparator for nonspecific behavioral effects of ketamine. Only the Investigational Drug Service was aware of drug identity, and all study personnel, including investigators, anesthesiologists, raters, participants, and data analysts, were blinded to randomization order.
Despite the widespread use of midazolam as an active placebo in ketamine studies, the acute intrainfusion side effects may still distinguish ketamine, and study subjects may thus become aware of the investigational agent they receive. A subset of participants completed a guess form at the 2-hour postinfusion ratings, on which they were asked to choose “ketamine,” “midazolam—active control,” or “I don’t know.” In order to maintain blinding, efficacy raters were not present during infusions and until after the 2-hour ratings were completed (after the side effects of ketamine and midazolam had dissipated). Participants were asked to refrain from discussing any infusion details with these raters.
Outcomes
The primary outcome measure was depression symptom severity as assessed by the Montgomery-Åsberg Depression Rating Scale (MADRS) (
26) 24 hours after the infusion. One day postinfusion was chosen as the primary endpoint based on the adult literature concerning the time course of ketamine’s antidepressant effects (
11), as well as our experience with cases in which pediatric patients were treated with ketamine (
20). We also expected all acute side effects of ketamine and midazolam to have subsided by that time.
Secondary outcome measures included MADRS time course (measured at 1 day, 2 days, 3 days, 5 days, 7 days, 10 days, and 14 days) and the CDRS-R (measured at 1 day, 7 days, and 14 days). The MADRS was chosen a priori as the primary outcome measure because in adult ketamine trials, the MADRS shows greater sensitivity to the acute changes associated with ketamine (
27) as compared to the Hamilton Depression Rating Scale (on which the CDRS-R is based [
23]). The effect size on a given day was calculated as the mean difference between ketamine and midazolam, divided by the square root of the pooled standard deviation (i.e., √(((SD
ket)
2 + (SD
mid)
2) / 2).
Acute side effects and any possible psychotomimetic or dissociative side effects were assessed with the CADSS. The hemodynamic effects of ketamine and midazolam were assessed by serial blood pressure, heart rate, respiratory, and oxygen saturation measurements.
Statistical Analysis
The primary analysis compared day 1 MADRS score after ketamine infusion and day 1 MADRS score after midazolam infusion via paired t test. This analysis included the 16 participants who received both study treatments. A sensitivity analysis with an unpaired t test was performed to include the whole study sample (N=17). MADRS and CDRS-R time course analyses were conducted for all 17 adolescents who participated in the trial. The planned sample size of 18 participants randomly assigned to treatment order was estimated to provide 80% power to detect a treatment effect size of 1.0 at 24 hours after infusion, based on effect sizes in adult studies (
11).
Carryover effects (i.e., whether treatment effects from the first infusion persisted into the second infusion period) were tested by comparing whether the difference in baseline scores between the first and second infusion periods were significantly different based on initial treatment assignment. Period effects (i.e., whether participants differed at the start of the first infusion compared with the second infusion regardless of treatment assignment) were tested by a paired t test comparing baseline MADRS scores in the first and second infusion periods regardless of initial treatment assignment. The presence of order effects (a treatment-by-period interaction) was determined via a log likelihood ratio test by systematically specifying nested models. Both nested models included first-order treatment and period effects. We defined a nesting saturated model with an additional treatment-by-period interaction term and compared it to a nested base model without the interaction term.
Dichotomous outcomes such as treatment response were analyzed using the McNemar test. Treatment response was defined a priori as a reduction >50% in MADRS score at any assessment conducted by the blinded rater within the 3 days following infusion. We defined response a priori as being present at any time between days 1 and 3 after infusion because adult study subjects typically experience their maximum response to ketamine somewhere between 1 and 3 days, and we wanted to capture all possible responders (e.g., participants who have an improvement on day 1 who do not meet response criteria, but further improve and meet response criteria on day 2) and to maximize statistical power.
In the mixed-model analysis of time course data, linearity was assessed graphically using scatterplots of the observed data and residual plots from linear regression models. In addition, linearity was evaluated analytically using polynomial models (linear, quadratic, cubic, quartic) and fractional polynomials. Graphical and analytic evaluation of linearity suggested an inflection point at day 1 (i.e., a spline with different slopes before and after the inflection point). Linear piecewise regression showed a larger decline in depression scores (both MADRS and CDRS-R) from baseline to day 1. After day 1, a different slope was present for the day 2 through day 14 time points. Therefore, a single linear relationship could not be assumed. To obtain different slopes for these different parts of the data, we centered the fixed part of the time variable (day of infusion) at day=1 in all mixed models and estimated the treatment-by-time interaction before and after day 1. Day 1 was also the time point when ketamine had its maximum effect. These models estimated fixed effects for treatment, time, period, and carryover effects. All tests were two-sided with alpha set at 0.05. Multiple comparisons are inherently considered with random intercept models. CADSS score, heart rate, and blood pressure side effect time courses on infusion day were analyzed via analysis of variance with repeated measures, with Bonferroni correction of comparisons of intrainfusion time points to baseline measurements.
Results
Participants
Of the 26 adolescents who were assessed for eligibility, seven did not meet inclusion criteria and the remaining 19 consented to participate in the trial (see the flowchart in the online supplement). Of the 19 who consented, 17 went on to receive the first infusion; one participant left the trial because of an undisclosed medical condition, and the other had a panic attack prior to starting the infusion pump for the first treatment and decided to withdraw before receiving the infusion. Sixteen of the 17 participants completed both infusions. One participant improved substantially after the first infusion and dropped out of the trial in order to receive ketamine treatment in the community. After the conclusion of the trial, this participant was confirmed to have received ketamine in the trial.
The demographic and clinical characteristics of the 17 randomized trial participants are summarized in
Table 1. All participants met criteria for major depressive disorder diagnosed via the K-SADS, and their mean screening scores were 63.2 (SD=17.1) on the CDRS-R and 33.1 (SD=9.3) on the MADRS. The average age was 15.5 years, and all ages between 13 and 17 years were represented in the sample. All participants had failed to respond to at least one 8-week trial of a standard antidepressant at therapeutic dosing, and on average the sample had 3.24 (SD=1.9) failed prior antidepressant treatments and 6.1 (SD=5.5) total psychiatric medication treatments, excluding ADHD medications (stimulants or alpha-2 agonists). The median number of ineffective antidepressant treatments was 2, with a range of 1 failed trial (N=2) to 7 failed trials (N=2). The average duration of the current depressive episode was 21 months (SD=18.8, median=12), reflecting a relatively chronic disorder for a pediatric population. Forty-seven percent of the sample (N=8) had a previous suicide attempt and 59% percent (N=10) had a history of nonsuicidal self-injury. None of the participants in the study were deemed to be imminently suicidal, in accordance with the exclusion criteria.
Participants remained on their current medication regimens during the trial, with nine participants (52.9%) on an SSRI, seven (41.2%) on a non-SSRI antidepressant, one (5.9%) on both an SSRI and a serotonin-norepinephrine reuptake inhibitor, and two (11.8%) on no medication. Five participants (29.4%) were on antipsychotic augmentation, one (5.9%) was taking lithium, and two (11.8%) were being treated with another mood stabilizer.
A guess form was completed by participants for 63% of all study infusions. Of 10 midazolam infusions, five participants correctly guessed midazolam (50%), two incorrectly guessed ketamine (20%), and three checked “I don’t know” (30%). Of 10 ketamine infusions, all 10 correctly guessed ketamine.
Difference in MADRS Ratings of Depression Severity 1 Day After Infusions
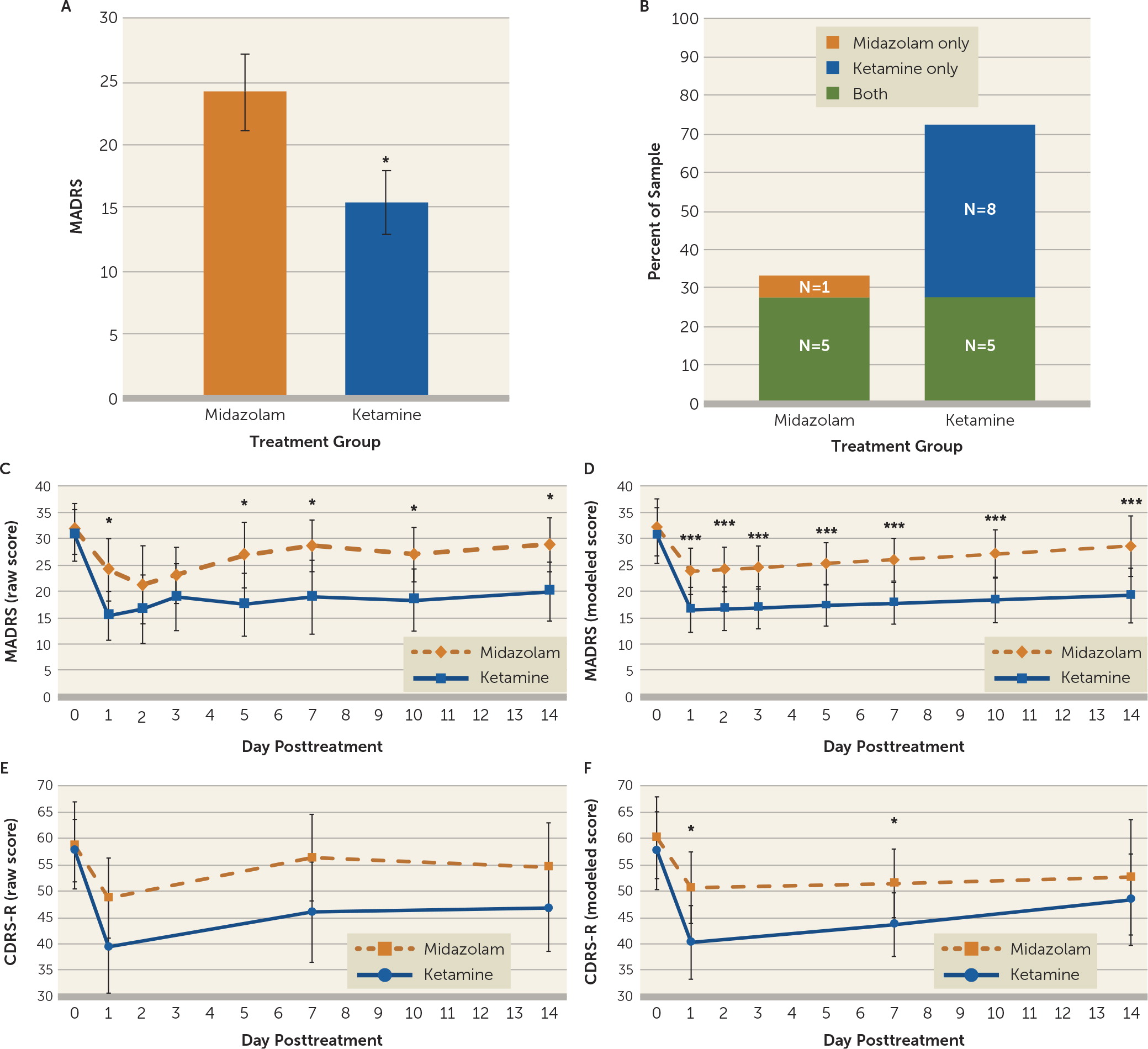
The primary outcome measure for the study was the MADRS score at day 1, comparing ketamine to midazolam. Ketamine significantly reduced MADRS scores compared with midazolam at day 1 after infusion (midazolam: mean=24.13, SD=12.08, 95% CI=18.21, 30.04; ketamine: mean=15.44, SD=10.07, 95% CI=10.51, 20.37; mean difference=−8.69, SD=15.08, 95% CI=−16.72, −0.65, df=15, p=0.036; effect size=0.78) (
Figure 2A). Additional sensitivity analysis that included the participant who received ketamine and then dropped out of the study, via an unpaired t test, yielded similar results (midazolam: mean=24.13, SD=12.08, 95% CI=18.21, 30.04; ketamine: mean=15.41, SD=9.75, 95% CI=10.78, 20.05; mean difference=−8.71, SD=10.94, 95% CI=−16.48, −0.94, df=31, p=0.029).
No significant carryover effects were observed in the trial. Potential carryover effects were tested by examining the difference between baseline scores in the first and second infusion periods based on whether participants received midazolam or ketamine first (d=2.32, 95% CI=−10.0, 14.73; t=0.40, df=15, p=0.69) (see the online supplement). A significant effect of period was observed, with participants showing reduced baseline MADRS scores at the beginning of the second infusion period compared to the beginning of the first infusion period regardless of initial treatment assignment (d=5.81, 95% CI=0.17, 11.61; t=2.14, df=15, p=0.025). Analysis using the log likelihood ratio test demonstrated no significant order effects (treatment-by-period interaction, p=0.40).
For the 16 participants who completed both phases of the study, the mean MADRS score at preinfusion baseline was 31.88 (SD=9.82, 95% CI=27.06, 36.69) prior to midazolam and 30.56 (SD=10.63, 95% CI=25.36, 35.77) prior to ketamine. The mean difference between treatment arms at baseline was −1.31 (SD=8.73, 95% CI=−7.89, 5.27, df=15) and was not statistically significant (p=0.68).
Proportion of Responders
Individual participants were assessed for significant clinical responses to ketamine or midazolam, which were defined as a reduction >50% in MADRS score within the 3 days following treatment (
Figure 2B). Participants were significantly more likely to respond to ketamine than to midazolam (McNemar χ
2=4.0, df=1, p=0.046); eight participants responded to ketamine only, compared with one participant who responded to midazolam only. Seventy-seven percent of the sample had a significant response to ketamine, which comprised eight participants who responded only to ketamine and five who demonstrated a response to both ketamine and midazolam. Thirty-five percent of the sample responded to midazolam, comprising the five participants who responded to both infusions and a single participant who responded only to midazolam and not to ketamine. Of the five participants who responded to both medications, one had received ketamine first and four had received midazolam first. Three participants did not respond to either infusion. Similar results were observed when the analysis of responders was restricted to day 1 after infusion (see the online supplement).
Model of Time Course of Treatment Effects: Regression Analysis of MADRS Time Course With a Linear Spline at Day 1
Figure 2C depicts the actual average MADRS scores in the ketamine and midazolam conditions during the course of the trial and
Figure 2D depicts the model examining these data over time. Consistent with our primary analysis, the modeled MADRS depression scores at day 1 following ketamine infusion were significantly improved compared with midazolam (midazolam: mean=23.85, 95% CI=19.46, 28.25; ketamine: mean=16.54, 95% CI=12.27, 20.81; mean difference=−7.32, 95% CI=−10.83, −3.80, p<0.001). Additionally, MADRS ratings after ketamine infusion were significantly lower than ratings after midazolam infusion at each postbaseline time point (days 1, 2, 3, 5, 7, 10 and 14).
In constructing the linear mixed model, a single linear relationship could not be assumed, as the change in depression symptoms differed markedly before and after day 1 (i.e., participants improved from infusion day to postinfusion day 1 and then slowly worsened toward baseline on days 2–14). Thus, the data were modeled in two phases: slopes at or before day 1, and slopes after day 1. In phase A (infusion day to postinfusion day 1), the difference in slopes comparing ketamine and midazolam effects (i.e., the treatment-by-time interaction) was −5.77 MADRS points per day (95% CI=−12.42, 0.89; p=0.089), which favored ketamine but did not reach statistical significance. Participants in both infusion arms exhibited significant improvement in phase A (infusion day to day 1): for the ketamine condition, MADRS score change was −14.11 points per day (95% CI=−18.72, −9.51; p<0.001), and for the midazolam condition, it was −8.35 points per day (95% CI=−13.15 to −3.54; p<0.01).
In phase B (postinfusion days 1–14), there was also no significant difference in slopes (i.e., treatment-by-time interaction) when comparing ketamine to midazolam, with the slope difference of −0.16 MADRS points per day (95% CI=−0.77, 0.46, p=0.62) visualized as parallel trajectories for the two medication conditions (
Figure 2D). From day 1 to day 14, the slope of change in MADRS score for ketamine was 0.21 points per day (95% CI=−0.19, 0.61) and for midazolam, 0.37 points per day (95% CI=−0.10, 0.83). Both groups maintained some of their symptom improvement during postinfusion days 2–14 but worsened compared with their initial improvement on day 1.
Differences in CDRS-R Ratings of Depression Severity After Infusions
Paired t test of CDRS-R scores on day 1 showed a nonsignificant reduction of scores with ketamine (midazolam: mean=48.38, SD=15.52, 95% CI=39.95, 56.82; ketamine: mean=40.54, SD=16.99, 95% CI=31.30, 49.77; mean difference=−7.85, SD=23.62, 95% CI=−20.71, 5.02, df=12, p=0.21; effect size=0.48). Independent t test on day 1 with all 17 participants produced similar results (midazolam: mean=48.75, SD=15.51, 95% CI=41.15, 56.35; ketamine: mean=39.50, SD=16.78, 95% CI=30.71, 48.29; mean difference=−9.25, SD=16.12, 95% CI=−21.33, 2.83, df=28, p=0.13). Similar to the MADRS analysis, there was no evidence of significant carryover effects. For the 16 participants who completed both infusion periods, the average CDRS-R score at preinfusion baseline was 58.75 (SD=16.86, 95% CI=50.49, 67.01) prior to midazolam and 57.38 (SD=12.78, 95% CI=51.11, 63.64) prior to ketamine. The mean difference of −1.38 (SD=16.16, 95% CI=−7.24, 9.99, df=15) was not statistically significant (p=0.74).
Figure 2E depicts the actual CDRS-R scores in the ketamine and midazolam groups during the course of the trial, and
Figure 2F depicts the model examining these data over time. Similar to the trajectory for MADRS ratings, a single linear relationship could not be assumed, as the change in CDRS-R scores differed markedly before and after day 1 (i.e., participants’ scores improved from the infusion to postinfusion day 1 and then slowly worsened toward baseline on days 2–14). Thus, data were again modeled in two phases: slopes at or before day 1 and slopes after day 1. In phase A (infusion day to postinfusion day 1), the difference in slopes comparing the effect of ketamine to the effect of midazolam (i.e., the treatment-by-time interaction) was −8.43 CDRS-R points per day (95% CI=−20.41, 3.54; p=0.17), which was not statistically significant. Both infusion groups exhibited significant improvement in phase A: after ketamine, participants experienced a change of −18.09 CDRS-R points (95% CI=−26.48, −9.70; p<0.001) from the infusion day to day 1, whereas after midazolam, participants experienced an improvement of −9.66 CDRS-R points (95% CI=−18.17, −1.15; p=0.026).
In phase B (postinfusion days 1–14), there was also no significant difference in slopes (i.e., a treatment-by-time interaction) when comparing ketamine to midazolam (0.51 CDRS-R points per day, 95% CI=−0.78, 1.82 p=0.44) (
Figure 2F). From day 1 to day 14, the slope of change in CDRS-R score for ketamine was 0.68 CDRS-R points per day (95% CI=−0.14, 1.49), and for midazolam, 0.16 CDRS-R points per day (95% CI=−0.83, 1.15).
Adverse Events
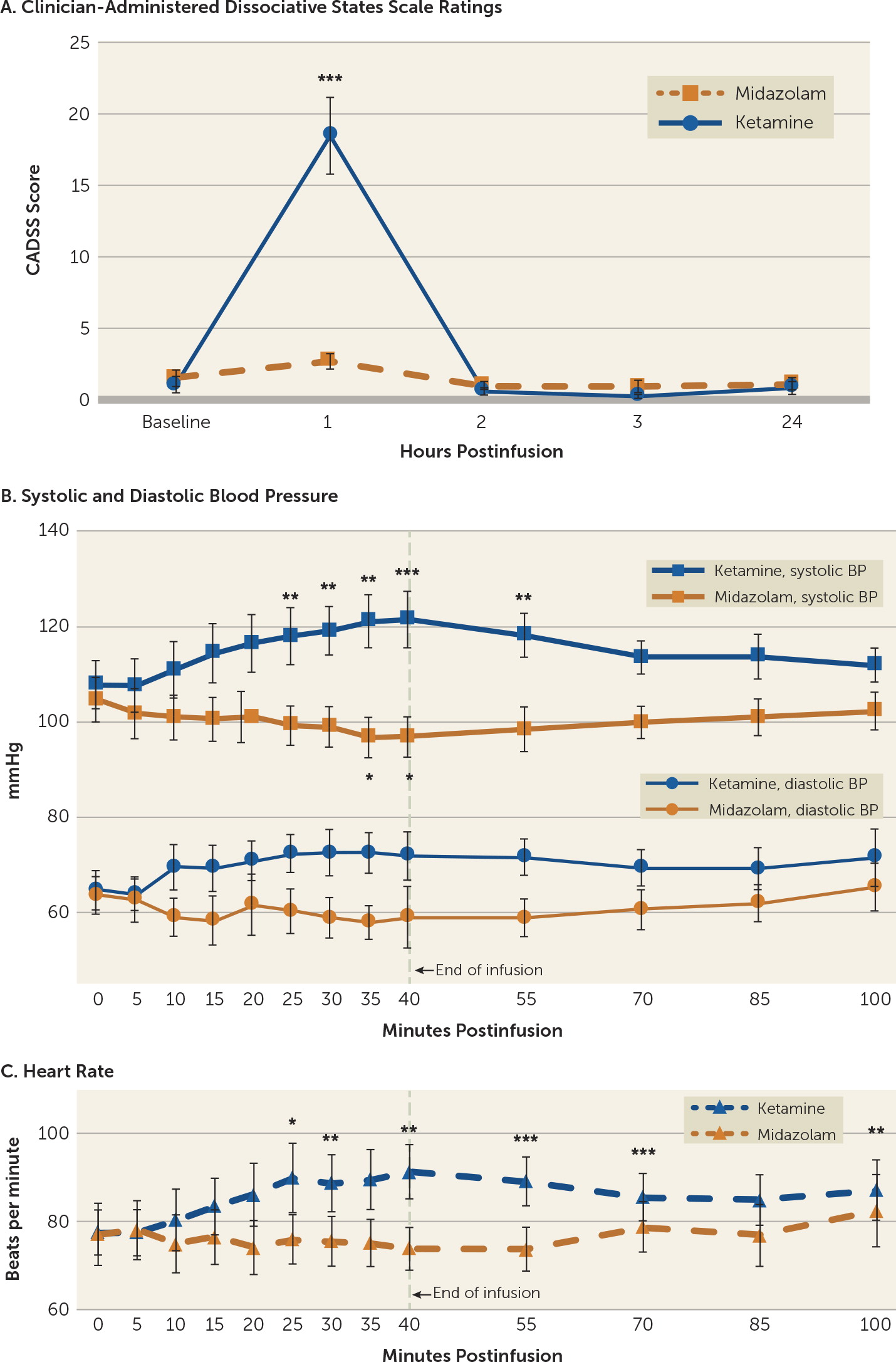
Significant dissociative symptoms, as assessed with the CADSS, were observed with ketamine treatment, although they were time limited (
Figure 3A) and participants were able to tolerate them. CADSS scores showed a significant effect of drug treatment (F=25.38, df=1, 15, p<0.001), time (F=12.93, df=3, 13, p<0.001), and the interaction of time with treatment (F=14.14, df=3, 13, p<0.001). Total CADSS score was elevated at the 1-hour time point (20 minutes after the end of the 40-minute infusion) and returned to baseline by the 2-hour time point (before the blinded rater conducted additional assessments). All 17 participants had a CADSS score >0 after ketamine infusions (range=1–39, median=17), and 14 of 16 participants (87.5%) had a CADSS score >0 after midazolam infusions (range=0–7, median=2). No significant dissociative symptoms were experienced in the days following the treatment. Aside from the participant who withdrew just prior to receiving infusion 1 because of anxiety (see the flowchart in the online supplement), none of the participants experienced significant dysphoria or panic, and none required emergent diazepam use. The most commonly reported dissociative side effects for each treatment condition are listed in
Table 2.
Systolic blood pressure was significantly influenced by drug treatment (F=23.08, df=1, 31, p<0.001), time (F=2.34, df=12, 372, p<0.007), and the interaction of time with treatment (F=13.19, df=12, 372, p<0.001). Diastolic blood pressure did not show an overall effect of time (F=1.26, df=12, 372, p=0.244), but it did show an effect of drug (F=14.98, df=1, 31, p=0.001) and an interaction of drug with time (F=3.47, df=12, 372, p<0.001). When treatment conditions were analyzed separately, ketamine-treated participants showed a significant effect of time for systolic (F= 9.91, df=12, 192, p<0.001) and diastolic blood pressure (F=3.08, df=12, 192, p=0.001). Systolic blood pressure significantly increased relative to baseline at 25, 30, 35, 40, and 55 minutes after the start of the 40-minute infusion, and diastolic blood pressure showed increased values at 35 minutes, although this fell short of significance. During midazolam infusions, systolic blood pressure was significantly influenced by time (F=4.21, df=12, 180, p<0.001), but diastolic blood pressure fell short of statistical significance (F=1.73, df=12, 180, p=0.064). Four participants had blood pressures meeting criteria for adolescent stage 2 hypertension (140/90 mmHg) (
28) during the ketamine infusion, but none persisted past the end of the infusion and none exceeded 150/95 mmHg. Midazolam infusions resulted in reduced systolic blood pressure relative to baseline at 35 and 40 minutes after start of infusion, and produced no significant changes in diastolic blood pressure.
Heart rate showed an overall effect of time (F=3.45, df=12, 372, p<0.001), drug (F=5.49, df=1, 31, p=0.026), and the interaction of drug with time (F=5.51, df=12, 372, p<0.001). When analyzed separately, heart rate during ketamine infusions was significantly influenced by time (F=6.91, df=12, 192, p<0.001), with increased heart rate at 25, 30, 40, 55, 70, 100 minutes after start of infusion. No participant had a heart rate exceeding 120 beats per minute. Heart rate during midazolam infusions also showed a significant effect of time (F=1.97, df=12, 180, p=0.029), although no individual time points were significantly different from baseline.
Discussion
In this trial we demonstrated that a single dose of intravenous ketamine, compared with a psychoactive placebo, midazolam, rapidly reduced depressive symptoms, as measured by MADRS score, in adolescents with treatment-resistant depression. Furthermore, we showed that on average, ketamine separation from midazolam persisted through 14 days, the longest time point examined in this short-term efficacy study. Ketamine was associated with transient, relatively mild dissociative symptoms and hemodynamic changes, and all participants were able to tolerate and complete the infusions. There were no serious adverse events during the study, no instances of participant withdrawal because of posttreatment adverse events, and no cases of persistent dissociation, emergent mania, or psychosis. This trial provides promising initial data on the feasibility, preliminary efficacy, and safety of intravenous ketamine in adolescent depression. However, additional data on long-term safety and efficacy are needed before any recommendations regarding integration into care in non-research pediatric populations can be made.
While case reports (
18,
20) and a recent open-label trial (
21) have generated increased interest in ketamine’s antidepressant potential in the adolescent population, this study represents the first placebo-controlled trial in adolescents. The sample was largely patients with treatment-resistant depression (
6) (averaging just over three failed prior antidepressant trials) and with relatively chronic (an average episode duration of 21 months) and severe symptoms (an average baseline CDRS-R score of 63.2, similar to the average baseline CDRS-R score of 59 in the Treatment of Resistant Depression in Adolescents [TORDIA] trial [
29]). Although placebo response rates may be lower in treatment-resistant populations (
30), a significant placebo response was evident in this study, as is common in pediatric depression trials (
22). The day 1 effect size of 0.78 in this adolescent sample is smaller than that reported in adult ketamine studies as a whole (effect size of 0.9 [
31]) but is in line with adult ketamine studies that use midazolam rather than saline as the control (effect size of 0.7 [
25]). At the group level, MADRS scores decreased on days 2 and 3 following midazolam infusion, despite being a short-lived improvement, whereas average improvement after ketamine infusion persisted through the 2-week period (
Figure 2C). At the individual level, 35% of participants met response criteria after midazolam, although the majority of these participants also had a significant response to ketamine (
Figure 2B).
Limitations of this proof-of-concept study include the small sample size and the relatively unbalanced randomized treatment assignments, which limited the power to detect carryover effects and order effects (treatment-by-period interactions). It will be important to replicate these findings in a larger, longitudinal parallel-design trial to better understand the durability of clinical response to both ketamine and the active placebo.
While the use of an active placebo is an important strength of our trial design, particularly relative to a saline control (
25), greater dissociative symptoms were reported following ketamine compared with midazolam (
Figure 3A). Additionally, hemodynamic measurements (blood pressure and heart rate), which are necessary to monitor for safety, were also significantly different at several time points between the two conditions (
Figure 3B). To help preserve blinding in light of these intrainfusion dissociative and hemodynamic differences, we utilized separate efficacy and safety raters and had efficacy raters who were not present at infusions (
22). However, this rater separation does not preclude the possibility of functional unblinding of the participants. Indeed, the withdrawal of one participant after ketamine infusion to pursue community ketamine treatment and the responses on a postinfusion guess form by a subset of participants suggest that functional unblinding likely occurs to some extent. While we cannot rule out the possibility that treatment expectancy plays a role in mood responses, this limitation applies broadly to all ketamine clinical trials, and midazolam is generally accepted to be the best available active control for ketamine at this time.
An additional potential limitation is the lack of separation of ketamine from midazolam at day 1 on the CDRS-R, which is the standard pediatric instrument in depression trials. The CDRS-R, however, was developed with the slower time course of conventional antidepressant–driven recovery in mind (
32), as opposed to the rapid action of ketamine and other rapidly acting agents. A significant portion of CDRS-R questions relate to observations spanning days and weeks (e.g., performance in school, acceptance or initiation of social engagements), which are difficult to adapt to a 24-hour time frame, especially when children in the trial did not typically go to school or see friends during the first day following the infusion. Thus, there may be reduced ability to detect signals of rapid change within this scale. As adult studies suggest that repeated dosing may extend ketamine’s duration of effect (
33), the CDRS-R may be a more appropriate instrument for future studies of repeated dosing that examine longer time frames. Overall, the CDRS-R results largely mirrored the MADRS data, with slightly reduced effect sizes.
An additional limitation is the sole use of structured rating scales and the absence of more patient-centered functional outcomes, which may better reflect meaningful depression recovery and do not always correlate with clinical symptom rating scales (
34).
The biological effects of ketamine are thought to relate to enhanced glutamatergic signaling, both via NMDA antagonism of prefrontal GABAergic interneurons (
35) and stimulation of AMPA receptors via enhanced glutamate release (
36) or ketamine metabolites (
37). The adolescent brain is a unique pharmacologic substrate, with active maturation of monoaminergic (
38), glutamatergic (
16), and GABAergic (
39) systems, and thus it is important to consider developmental pharmacologic context in the development and testing of novel therapeutics. While ketamine has an excellent acute safety record in pediatric patients (
19), the risks of multiple exposures are not fully known. Animal studies suggest unique vulnerability of the developing brain to the neurotoxic effects of high-dose ketamine (
40), and studies of people with ketamine use disorder suggest potential deleterious neurocognitive effects (
41). While this initial proof-of-concept crossover trial provides early evidence suggesting positive short-term efficacy and safety of a single ketamine dose in this population, clinical treatment and future studies, particularly those that employ multiple-dosing strategies, should be approached with care and caution.