Case 1
A boy, age 2 years and 8 months, presented with seizures characterized by sudden onset of unstable gait, accompanied by left limb hypodynamia in the morning. Several minutes after each seizure, the child experienced a unilateral oral twitch and deviation of head and eyes, which were associated with a secondary generalized seizure. The seizures, which occurred several times daily, decreased in severity after an intramuscular injection of phenobarbitone (PB) and were seen only intermittently (once every few days) thereafter.
Two months after PB treatment, the patient fell suddenly, exhibiting hemiconvulsion, posthemiparesis, and a positive Babinsiki’s sign on the left side of his body. Various symptoms of seizures were then seen, including oral twitching and staring, as well as signs of hemiclonic seizure. On several occasions, the patient demonstrated unilateral status epilepticus (SE), gradually progressing to epilepsia partialis continua (EPC), which was gradually transformed into intermittent focal clonus. He then responded to intravenous midazolam. Since discharge from the hospital, he has complained of repeated somatosensory symptoms, and experienced a focal seizure and drop attack on the left side of his body, despite continuous oral administration of carbamazepine (CBZ) 300 mg/day and prednisolone (PDS) 20 mg/day.
By Month 11 after the onset of illness, the patient still exhibited focal seizures, along with unilateral neurological and cognitive deficits, although not as severe as before. After topiramate 75 mg/day po was added, a striking improvement in seizure control and cognition was observed. However, the patient still experienced hemiparesis and occasional drop attacks.
During the first onset of illness, immunological tests of his serum were positive for cytomegalovirus (CMV) immunoglobulin (Ig) (CMV-IgG). Chemistry and virology tests of the patient’s cerebrospinal fluid were normal, but IgM (0.069 mg/dL) was markedly higher than normal (0.0011–0.0370 mg/dL).
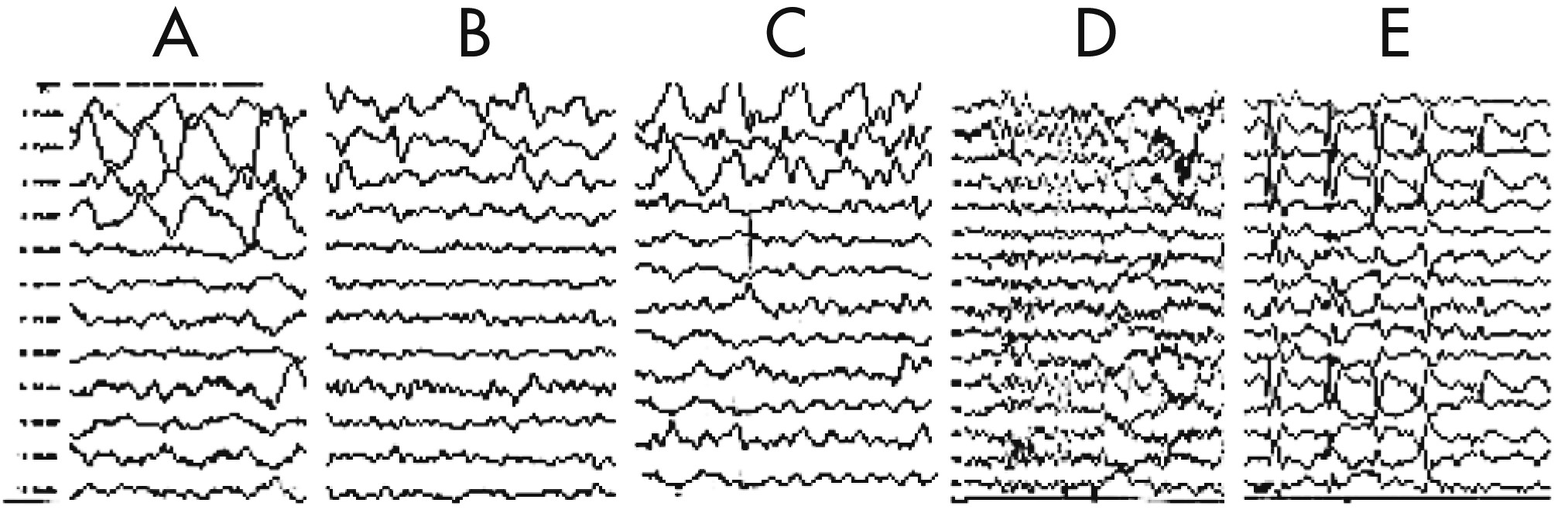
The EEG taken during the initial stage of disease revealed diffuse, high-amplitude, polymorphic delta activity over the affected hemisphere, especially over the right frontal lobe. This activity shifted to the left frontal region for a short time (1 month) and became a distinctive early feature of his condition (
Figure 1 [A–C]). After 9 months, EEG revealed progressive impoverishment of background activity over the affected hemisphere and dominant spike-wave activity over the right frontotemporal area during sleep (
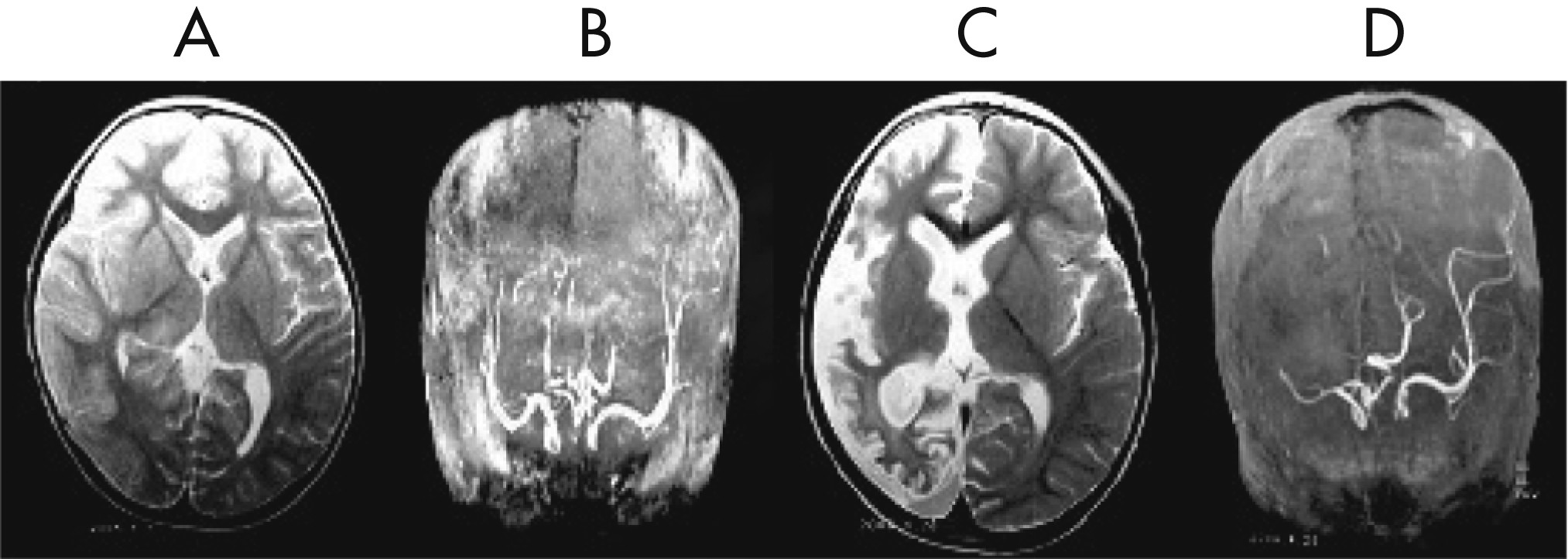
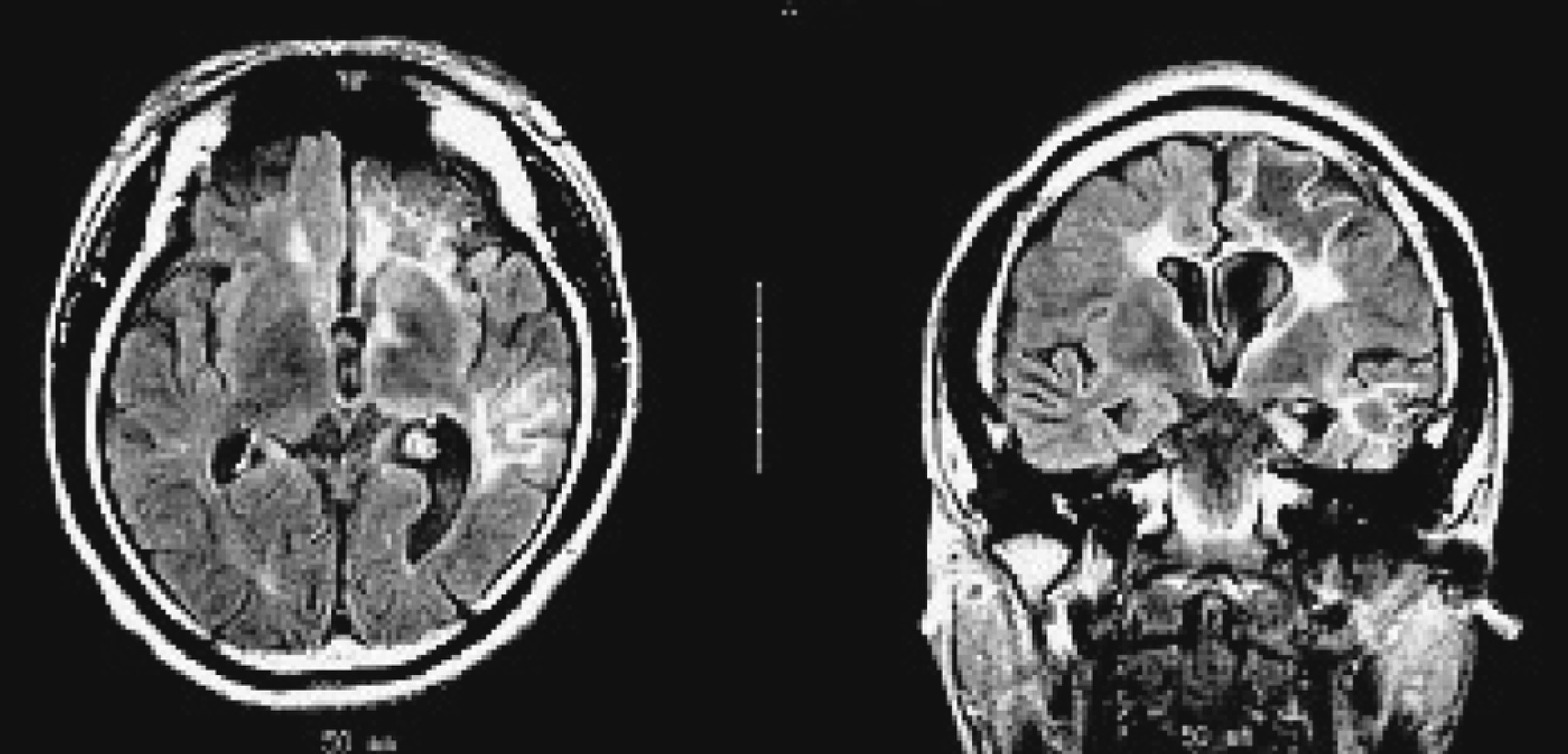
Figure 1 [D, E]). His initial MRI revealed an obvious focal area consisting predominantly of edema coinciding with a depression in the ipsilateral ventricle. The hemispheric ratio (HR) was 1.26, which indicates the earliest stage of RE. Within 9 months, HR declined to 0.72, indicating a considerable amount of atrophy, and enlargement of the lateral ventricle on the involved side of brain (
Figure 2 [A, C])). The MRA revealed a distinctive feature of a later stage of the disease: a mild depression and shift in the location of the cerebral middle artery (CMA), progressing to a later stage characterized by the absence of the CMA and cerebral anterior artery (CAA;
Figure 2 [B, D]).
Case 2
A boy 6 years and 4 months old experienced a head injury during a fall. He then experienced a hemiconvulsion on the right side of his body that continued for about 10–20 minutes, accompanied by ipsilateral eye deviation, dysarthria, and transient postictal hemiparesis. The seizure recurred about 1–2 times per month, despite treatment with PB 90 mg/day. Five months later, the disorder became aggravated, characterized by frequent asymmetric clonic seizures, accompanied by eye deviation, speech deficit, and oral and manual automatism. These symptoms progressed to unilateral or generalized motor manifestations. During this period, he demonstrated a transient response to CBZ 500 mg/day and PB 90 mg/day. At age 6 years and 11 months, the patient still experienced illness progression, which was characterized by several types of seizures, including simple partial seizures, occasional EPC, or prolonged ipsilateral complex partial seizures or secondary generalized tonic–clonic seizure (sGTCS), associated with mental and neurological deterioration.
Neither CBZ 600 mg/day nor valproate (VPA) 800 mg/day were useful for this patient, so human intravenous immunoglobulin (IVIG) 400 mg/kg/day was given by infusion for 5 days. Seizure frequency decreased, and neurological function improved after 3 additional infusions of IVIG monthly. Because of intermittent partial seizures (PS), EPC, and sGTCS, PDS 40 mg/day was administered, along with oxcarbazepine (OXC) 600 mg/day and PB 90 mg/day. At the 1-year follow-up visit, seizures were markedly controlled, and mental and neurological functions were greatly improved. The patient still experienced PS and occasional drop attacks once daily or every several days, but refused surgery because of the risk of damage to speech functions.
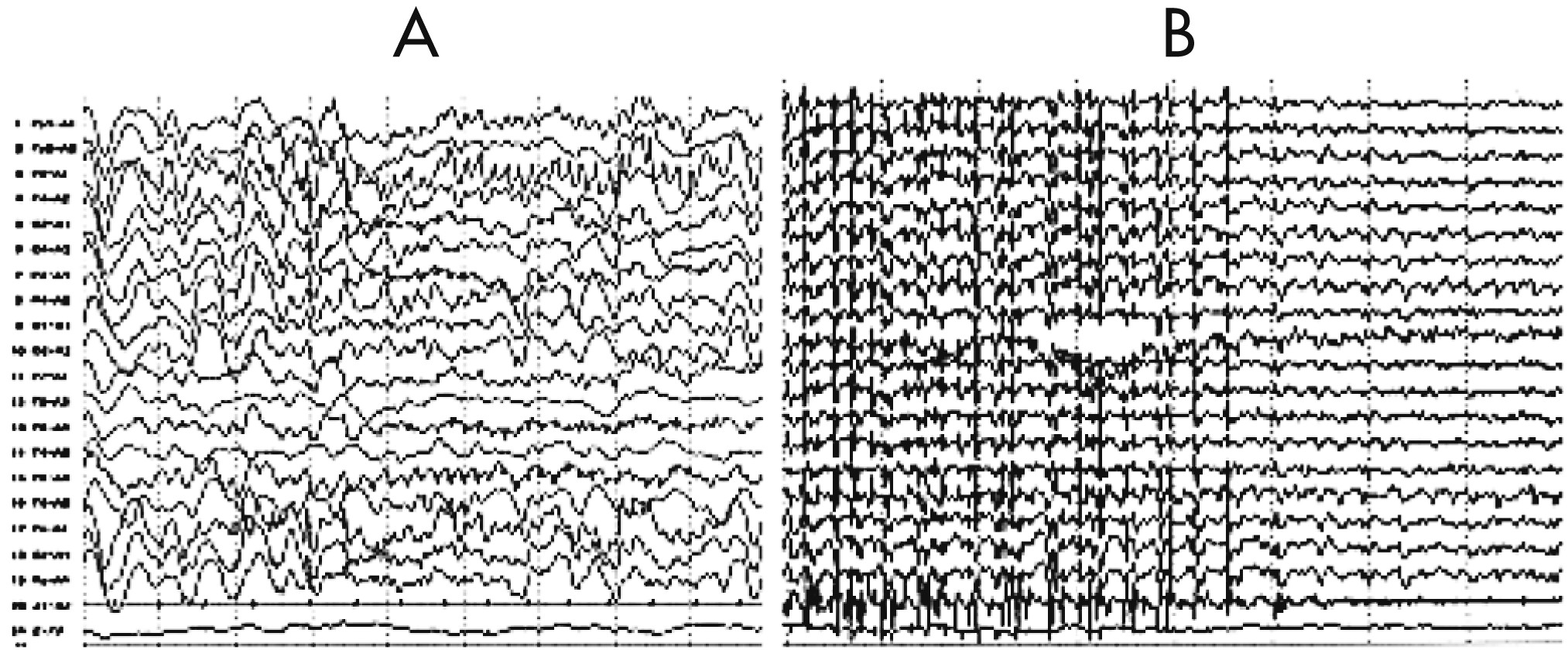
An EEG revealed that a diffuse slowing background interposed with focal rhythmic epileptic activity had spread over the entire brain in association with sGTCS (an early feature of RE;
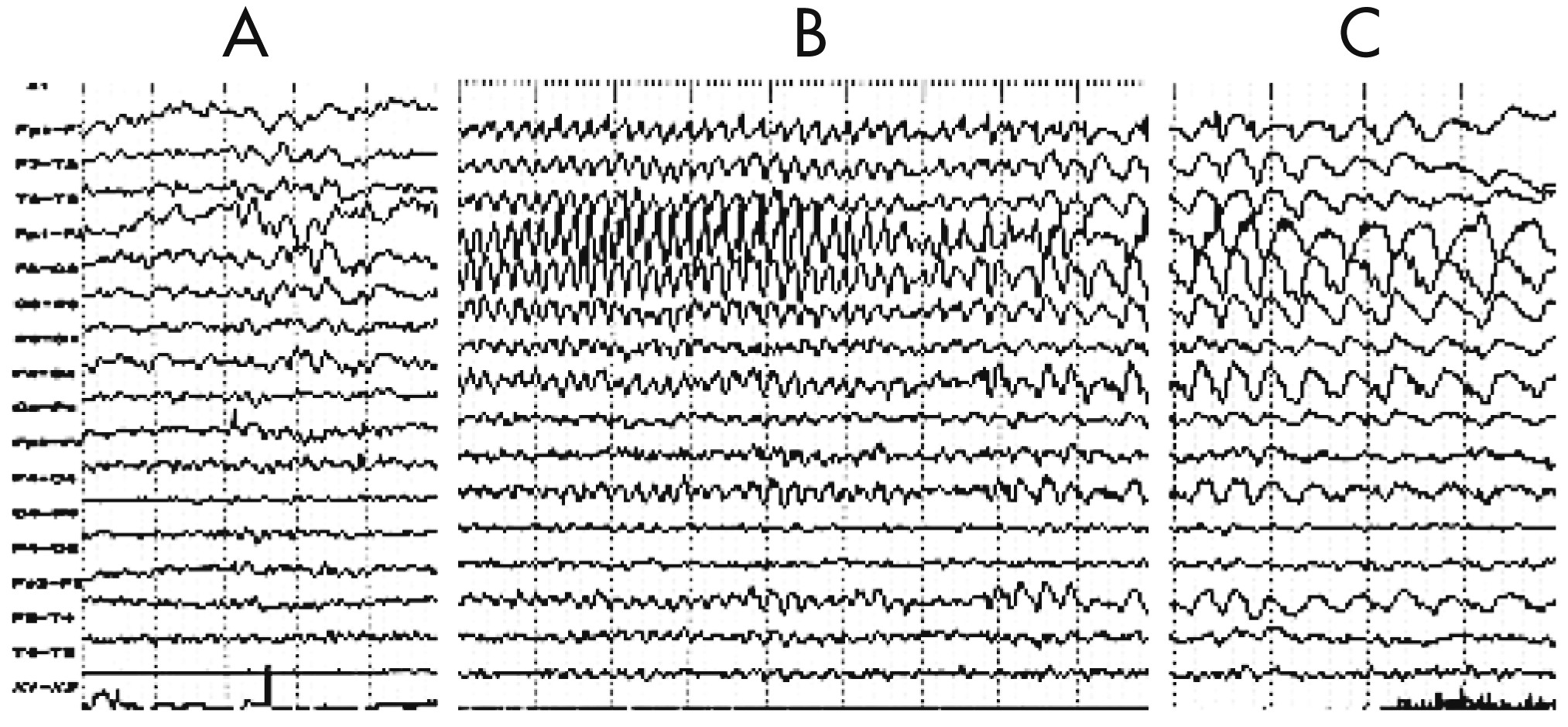
Figure 3). Five months later, EEG showed diffuse reduced activity, intermingled with epileptic abnormalities over the left frontal-temporal-parietal regions that coincided with PS, EPC, dysarthria, and automatism, and served as another distinctive feature of the disease (
Figure 4 [A–C]). The MRI revealed diffuse cerebral atrophy intermingled with focal atrophy accompanied by dilation of the left lateral ventricle of the brain (
Figure 5).