Case Presentation
A 68-year-old right-handed woman presented to our behavioral neurology subspecialty clinic with at least a 3-year history of cognitive decline; she had recently retired from her job as an administrative assistant. Symptom onset was insidious and initially characterized by the patient forgetting words and names of people in conversations. The patient was evaluated 1 year earlier by her primary care provider, who noted that she had to be “reminded of conversations,” which was in sharp contrast to her usual “highly organized” nature. After noticing cognitive difficulties, her supervisor at work encouraged her to retire. Over time, she began to ask questions repeatedly in conversations. By the time she was evaluated in the behavioral neurology clinic, only a very brief amount of time would elapse before she would repeat a question. While grocery shopping, she would pick out items from the shelves that she had already put in her cart.
The patient had a long-standing anxiety disorder, characterized by intermittent panic-like symptoms that did not meet DSM-5 criteria for panic disorder, and she had anxiety related to driving. In addition, she had a history of becoming very anxious in the context of psychosocial and health-related stressors. For example, she had debilitating anxiety during previous evaluations for possible breast cancer, even though there was low suspicion for malignancy. Her generalized anxiety disorder had been well controlled for several years with escitalopram. In the period shortly before her behavioral neurology evaluation, her anxiety had dramatically worsened. She started to avoid being alone in public places because she feared that she would get lost.
The patient drank alcohol throughout her adult life, starting with occasional use in her twenties and increasing in quantity with age: she drank two glasses of wine daily in her forties and three to four glasses by her late fifties. Consistent moderate to heavy alcohol use continued until her primary care physician advised her to reduce consumption. A few months before her behavioral neurology evaluation, she had successfully reduced her alcohol consumption to one drink daily, without notable cognitive improvement. She was also advised several times to undergo evaluation for obstructive sleep apnea (OSA), given the presence of mild obesity, loud snoring, and nocturnal apneic events, but she declined to follow through.
The patient did not report or exhibit any clear difficulty with basic attention or visuospatial function. She and her husband denied that she had other neurological symptoms, such as gait difficulty, muscle weakness, visual changes or misperceptions, vertigo, headaches, limb rigidity, fluctuating levels of arousal, sensory changes, or abnormal movements (e.g., tremors). She did not have any history of head injury or seizures.
Despite progressive worsening of her cognitive symptoms 3 years after the onset of symptoms, with deficits readily discernible to the observer, the patient and her husband reported overall independence in instrumental activities of daily living (IADLs), including cooking meals, driving, and shopping for groceries. She was aware of her cognitive changes and was concerned that her memory was impaired because of her long-standing anxiety and alcohol use.
Other medical history included hypertension, hypothyroidism as a complication of treatment for Graves’ disease, and hypercholesterolemia. She had been euthyroid for decades because of levothyroxine administration. In addition to levothyroxine, her medications included escitalopram, olmesartan, amlodipine, hydrochlorothiazide, and atorvastatin.
The patient was a high school graduate with no history of learning disabilities or developmental delay. She had a remote history of smoking two packs of cigarettes daily for 17 years. Her mother had a history of memory complaints in her early seventies and emphysema requiring oxygen therapy and anxiety. Her sister had anxiety and depression, and one maternal uncle had excessive alcohol use. There was no known family history of neurological or neurodegenerative disease.
Questions: What are the diagnostic considerations based on the history? How might a clinical examination help to narrow the differential diagnosis?
The patient had a gradual onset of progressive cognitive decline, occurring over at least 3 years and likely starting in her mid-sixties. Her primary symptoms suggested impairments in the cognitive domains of episodic memory and language, evidenced by rapidly forgetting new information and difficulties retrieving words and names. This history is most suggestive of an underlying neurodegenerative disease. This pattern of cognitive impairment based on her history is typically seen in the common, amnestic, multidomain variant of Alzheimer disease (AD) (
1,
2). Given her history of daily, moderate to heavy alcohol use for at least 10 years, her alcohol use was considered to be at least a contributing factor, if not one of the primary etiologies. Alcohol-related brain damage—a condition reflecting neurotoxic effects from ethanol, thiamine and other nutritional deficiencies, and liver dysfunction—can manifest as either an amnestic syndrome, with prominent anterograde and retrograde amnesia (i.e., alcohol-induced persistent amnestic disorder or Korsakoff syndrome), or a nonamnestic impairment, including executive dysfunction and impaired explicit memory and visuospatial function (
3,
4). Other contributors that were considered included her severe anxiety disorder, which may present with symptoms of executive dysfunction manifesting as forgetfulness (
5,
6), and her suspected OSA, because this can also contribute to forgetfulness (
7). Other differential diagnoses and potential contributors to her cognitive impairments included cerebrovascular disease—which has an increased incidence among patients with hypertension, hypercholesterolemia, obesity, and smoking history—and hypothyroidism-related cognitive impairment. Both vascular cognitive impairment and hypothyroidism can present with memory-retrieval difficulties and, occasionally, true amnesia in the case of vascular lesions affecting temporolimbic structures (
8,
9).
A detailed clinical examination can help clinicians evaluate for physical stigmata of alcohol use disorder, including pallor, icterus, asterixis, and withdrawal tremors in the hands and tongue. A neurological examination can reveal evidence of length-dependent symmetric peripheral neuropathy, subtle muscle wasting, gait dysfunction, or cerebellar signs, which all increase the likelihood that there are other central nervous system effects of alcohol. Evidence of neck nodules or gland enlargement can signal a thyroid disorder, and obesity, a thick neck, and hypertension would make OSA a more likely contributor. Evidence from the examination of prior stroke, including subtle hemiparesis with upper motor neuron signs, or other focal neurological findings can support contributions from cerebrovascular disease.
A neurobehavioral status examination, assessing cognitive functions such as verbal and visual memory, visuospatial function, attention, executive abilities, language, and praxis, as well as measurements of mood and affect, will also inform the differential diagnosis by confirming the profile and severity of cognitive deficits and providing additional information about the potential impact of anxiety on the patient’s presentation.
Our patient’s vital signs were normal. Her body mass index (BMI) was 29.8 kg/m
2, approaching the 30-kg/m
2 threshold for obesity (
10). Her neurological examination revealed depressed patellar reflexes and absent Achilles deep tendon reflexes bilaterally. She had significant difficulty providing any autobiographical information. She appeared highly anxious and overwhelmed during the interview, with almost panic-stricken and stunned facial expressions when she was asked basic questions. She could not provide her correct age and was not oriented to the year, date, or time of day. Her responses were terse and unsure, and she deferred to her husband for most answers. Only limited cognitive testing was conducted because of her overall level of anxiety and resistance to assessment. She struggled to complete the modified Trail Making Test, Part B, component of the Montreal Cognitive Assessment (MoCA) (
11), despite repeated instructions. She could not copy a wire cube or appropriately draw the arrayed numbers or hands on the MoCA clock-drawing test (CDT). Notably, on the CDT, she wrote in only numbers 1 through 9, which were unevenly spaced, and then she drew a line from each number to the center of the circle. She made three attempts to encode five words, and she could recall only one word after 5 minutes. She was able to recall the remaining four words with semantic cueing. Her husband rated her with a perfect score (8/8) on the Lawton-Brody Instrumental Activities of Daily Living Scale, suggesting no loss of independence in performing most IADLs. Notably, this scale does not assess for changes in function in occupational settings (an advanced aspect of ADLs), which she had exhibited.
Questions: How does the examination contribute to our understanding of diagnostic considerations? What additional tests or studies are indicated?
The patient’s elemental neurological examination was normal except for depressed lower-extremity deep tendon reflexes. This can be seen among individuals with obesity, older individuals, and individuals with length-dependent symmetric polyneuropathy, a complication of chronic alcohol use (
12,
13). A high BMI and the observation of apnea while she slept strongly pointed toward possible underlying OSA. She did not have any stigmata of hypothyroidism. Limited office-based cognitive testing indicated impairments in multiple cognitive functions, including memory encoding and delayed recall, aspects of executive function (e.g., set-shifting and planning), and aspects of visuospatial function (e.g., visual construction). This examination was not sufficient to establish the presence or absence of temporolimbic amnesia, particularly in the context of her history and other suggestive factors (e.g., poor autobiographical recall and impaired temporal orientation). Likewise, the examination could not confirm language deficits that could be assessed with tests of confrontation naming and verbal fluency. That said, the progressive time course, coupled with a cognitive profile involving episodic memory, language, executive, and visuospatial dysfunction suspected through both the history and the neurological examination, would be observed most commonly in the context of underlying AD neuropathology (
14).
The incongruence between her reported intact daily functioning and her poor cognitive testing performance could reflect variability in her performance across settings; alternatively, this incongruence could suggest that she may have been struggling more in day-to-day activities than was initially apparent and that she was on the cusp of losing independence. In addition, her profound anxiety throughout the examination, even when asked simple questions, likely contributed to her poor performance. Stress and anxiety may independently impair memory encoding, delayed word recall, and executive function by negatively affecting working memory (
15,
16). There is accumulating evidence that the emergence of anxiety, or worsening of chronic anxiety, may be prodromal to underlying neurodegenerative disease, reflecting a reciprocal relationship between anxiety and cognitive dysfunction (
17,
18).
We recommended laboratory tests, including complete blood count, comprehensive metabolic panel, thyroid function tests, vitamin B
12 status, and syphilis and HIV serologies, as part of a standard evaluation for non-neurodegenerative contributors to cognitive decline. Ideally, a polysomnogram to evaluate OSA would have been obtained. We also recommended a brain MRI to assess for structural changes or focal patterns of atrophy and vascular injury, which may have shed light on likely underlying pathologies. Given her symptoms of progressive memory loss, we might expect relative volume reduction in mesial temporal lobes compared with other regions, as seen in AD (
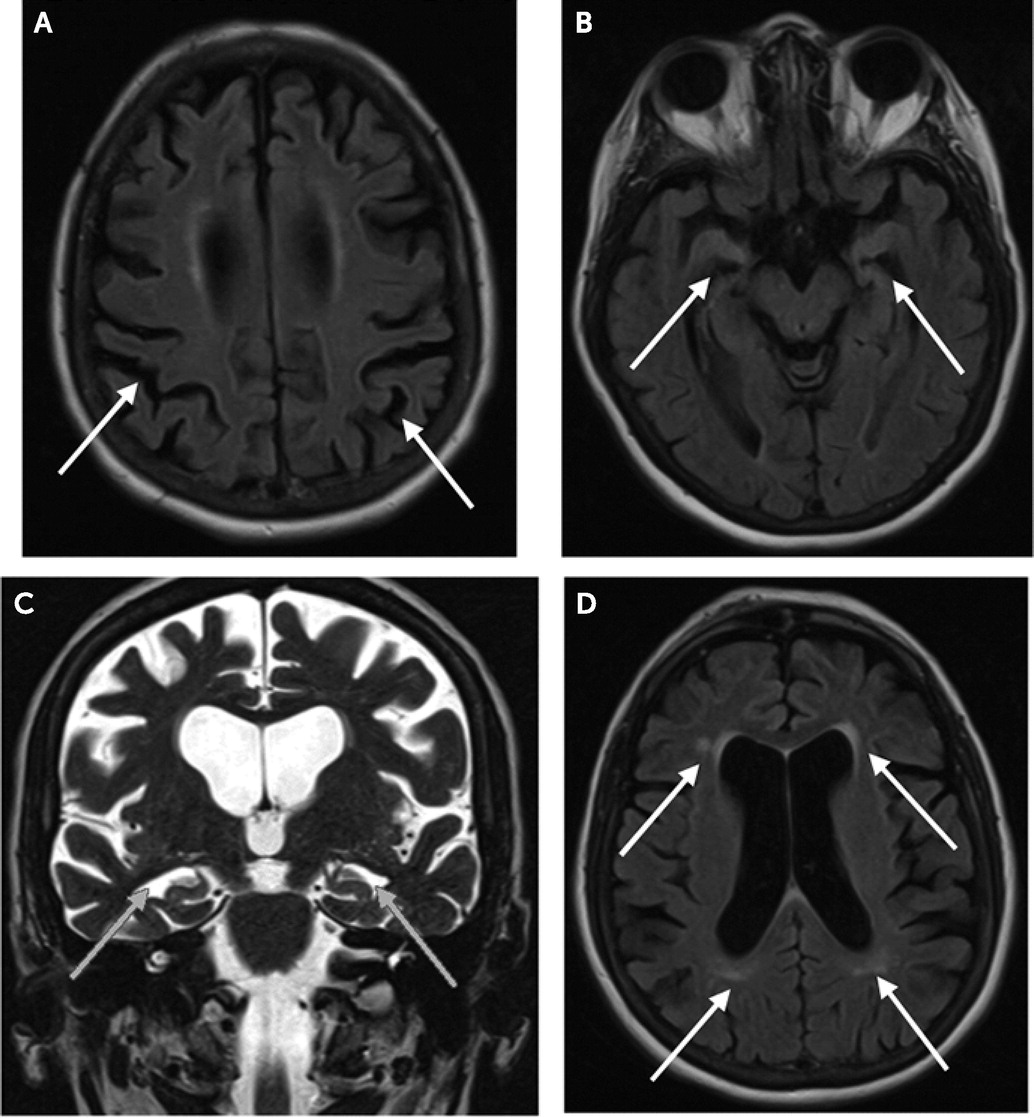
14). Her laboratory results were within normal limits. Brain MRI (
Figure 1) revealed mild, bilateral mesial temporal and posterior parietal volume loss and demonstrated mild, scattered T2 white matter hyperintensities in the deep white matter and periventricular region.
Amnestic impairment, coupled with evidence of disproportionate mesial temporal volume loss on MRI, can be helpful in predicting AD pathology, especially in cases like that of our patient, where obtaining cerebral spinal fluid (CSF) and positron emission tomography (PET)-based AD biomarkers is not feasible because of profound neuropsychiatric symptoms. Mesial temporal lobe atrophy is a sensitive marker for the diagnosis of AD and for the clinical progression from mild cognitive impairment (MCI) to dementia, where further volume reductions parallel worsening cognitive impairments (
19). Mesial temporal lobe structures, such as the hippocampus, entorhinal cortex, perirhinal cortex, and parahippocampal cortex, are vital for multiple cognitive functions, including declarative memory and spatial navigation (
20), and are the initial regions of neurofibrillary tangle accumulation in AD (
20). Hippocampal substructures lose volume at a significantly steeper rate among individuals with AD-related MCI who convert to dementia than among individuals with MCI who do not convert (
20). Importantly, mesial temporal lobe volume loss has also been linked to normal aging and non-AD neurodegenerative pathologies, and it may not be possible from imaging at one time point to differentiate normal age-related changes or non-AD changes from those that reflect early AD-related pathological change, particularly among older individuals (
21–
23). Interestingly, hippocampal and limbic-prefrontal circuits have been implicated in anxiety (
24), which our patient endured since her middle adulthood; the exacerbation of anxiety in her sixties may be at least temporally correlated to degeneration in these regions.
Question: Considering these additional data, what would be an appropriate diagnostic formulation?
Imaging results provided additional support for a diagnosis of a progressive amnestic (multidomain) syndrome likely associated with AD neuropathology with additional negative effects from generalized anxiety, prior heavy alcohol use, and possible OSA. While preservation of independence in IADLs and basic ADLs would place her at a level of MCI, grossly apparent deficits, history of occupational dysfunction leading to early job retirement, and poor performance on limited cognitive testing suggest that she may have been on the verge of losing independent functioning.
Question: How might contributions from alcohol and anxiety be considered more carefully?
We hypothesized that our patient’s prolonged, heavy alcohol use may have contributed to her cognitive impairment. Central nervous system effects of alcohol are more likely when there is evidence of other neurological or systemic injury. While she may have had a length-dependent peripheral neuropathy, no sensory examination or electrodiagnostic testing was completed to confirm this diagnosis, and this type of neuropathy is not specific to alcohol misuse (
3). The potential effects of alcohol on her cognition were difficult to establish with any certainty, given that cognitive symptoms are varied with alcohol-related brain damage, and there are no reliable biomarkers of alcohol-related cognitive impairment. Several features make it reasonable to argue against a significant component of alcohol-related cognitive impairment. There was a lack of supportive alcohol-related neurological examination findings (i.e., there were only suggestive neuropathic signs), including cerebellar signs, ocular signs, and ataxia (as might occur in Wernicke-Korsakoff syndrome), and lack of signs of hepatic encephalopathy. There was no imaging evidence of Wernicke-Korsakoff syndrome (e.g., T2 hyperintensities in the mamillary bodies, periaqueductal gray matter, and region around the third ventricle), Marchiafava-Bignami disease (e.g., atrophy and T1- and T2-signal changes in the corpus callosum), or alcohol-related cerebellar degeneration. Furthermore, she had no improvement in cognition despite a significant reduction in alcohol consumption over several months, which at least suggests that there was no evidence of short-term or additive effects of current alcohol consumption on her impairment, even if chronic alcohol use was contributory (
4,
25,
26).
The relationship between alcohol use and cognition has been debatable, with some evidence suggesting that light to moderate use is protective against dementia (
27,
28). However, a recent, large U.K. Biobank-based study found a negative dose-dependent association between alcohol use and both gray and white matter volume, even among healthy middle-aged and older adults who consume only one to two units of alcohol daily (
29). Relevant to our patient, another study found that older individuals (age ≥72 years) with MCI who exceeded 14 drinks per week (as she did) had the most severe cognitive deficits, compared with their peers with MCI who drank less (
30).
While our patient had a history of long-standing anxiety, her anxiety increased dramatically in the context of her cognitive decline. Late-life spontaneous psychiatric exacerbation may occur with or without a concurrent neurodegenerative process. Her worsening anxiety was likely multifactorial in etiology, with probable contributions from both disruption of brain circuits by neurodegenerative pathology and her emotional response to her cognitive impairment. The latter has been described with anxiety and other neuropsychiatric symptoms among individuals with neurodegenerative disease (
31,
32). We will discuss this topic further in the Discussion section. In addition to the independent effects of alcohol use and anxiety on cognition, alcohol misuse and anxiety also directly influence each other (
33).
Question: Does information about the longitudinal course of her illness alter the formulation about the most likely underlying neuropathological process?
Within 2 months of evaluation in the behavioral neurology clinic, our patient’s anxiety had worsened, and she developed paranoia in the context of her husband’s serious medical illness, hospitalizations, and untimely death. Her anxiety symptoms prompted an increase in her daily dose of escitalopram and the addition of mirtazapine as an adjunct. Her anxiety significantly improved with this medication regimen. On the other hand, her cognitive difficulties increased and daily functioning notably worsened during her husband’s hospitalizations and following his death. Within 1 year of being diagnosed with MCI, she started to have significant difficulty driving, indicating a clear progression to dementia. Her cognitive symptoms were treated with donepezil.
By 4.5 years after symptom onset, she had difficulty recognizing close family members, could not recall salient remote autobiographical details, and consistently confused the names of people and places. She had difficulty recalling her son’s full name when she heard only his nickname. Her family became concerned about her ability to live alone independently. After 6.5 years, she required assistance with even basic ADLs and moved into a long-term care facility. By this time, her speech output was reduced, and she had much difficulty adjusting to the new environment of the facility. She developed persecutory delusions, skin picking, wandering behavior, and restless sleep; these symptoms required the addition of risperidone and melatonin to her medication regimen, which helped reduce the symptoms and related distress. For the last 1.5 years of her life, she benefited from hospice care, and she died approximately 9 years from symptom onset.
Thus, our patient had a progressive amnestic-predominant loss of cognitive function and abilities over many years following her diagnosis. While not specific for AD, her delusions, agitation, wandering behavior, and paucity of speech are common with AD progression. The longitudinal course of her condition, when coupled with her initial syndrome and MRI findings, was consistent with our initial diagnostic impression of underlying AD neuropathology.
Discussion
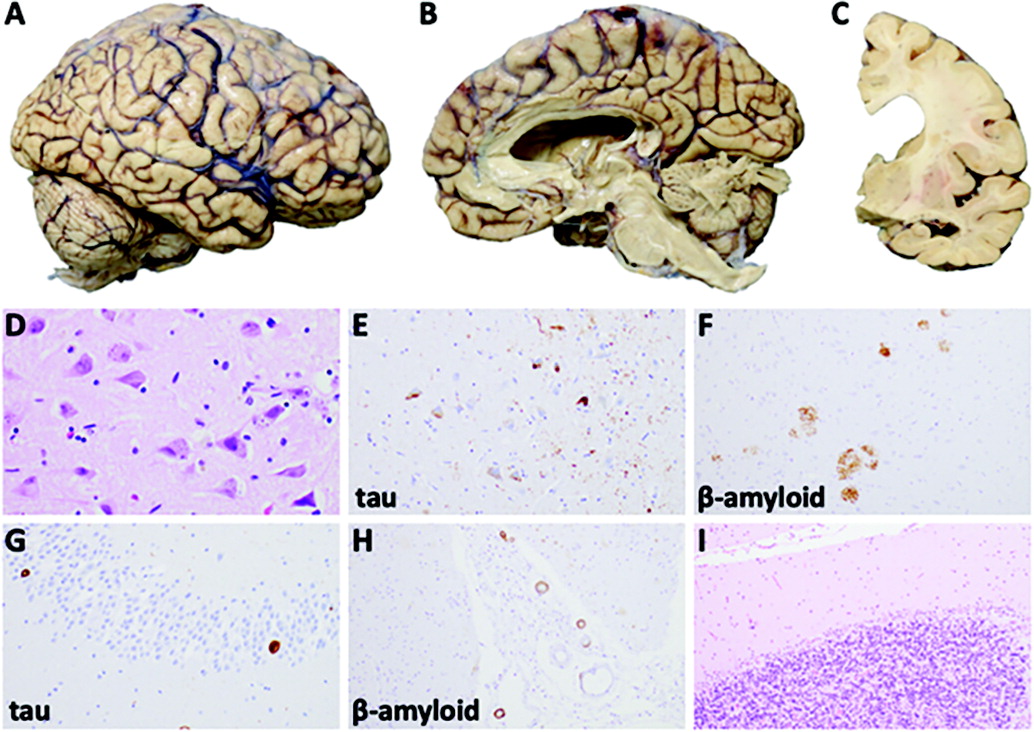
A patient presented in her late sixties with a syndrome common among those with AD. The syndrome was characterized initially by difficulties with episodic memory and aspects of language (including word retrieval), and these symptoms were observed in the context of severe anxiety, alcohol use, and likely OSA. By the time she was evaluated in a specialty clinic, she had already developed other cognitive deficits, including problems in aspects of executive function, autobiographical memory, working memory, and visuoconstruction ability. Within 1 year of diagnosis, she progressed from having late-stage MCI to clear mild dementia with prominent neuropsychiatric symptoms. Prolonged heavy alcohol use, the presence of multiple systemic vascular risk factors, and likely (but undiagnosed) OSA may have all contributed to the observed cognitive decline. Notably, only effects associated with AD and systemic vascular disease were evident on neuropathological examination. Antemortem clinicopathologic correlation predicted that AD would be the dominant underlying pathology, which was ultimately confirmed.
Some pertinent clinical features of this case include a clear exacerbation of long-standing anxiety concurrent with the onset and progression of cognitive dysfunction, as well as discordance between reported day-to-day functioning and objective cognitive performance during evaluation.
In a recent study, the probability of progression from MCI to mild AD over 1 year at age 65 was 22% (
35). We hypothesized that our patient’s cognitive and functional status declined rapidly, in part, because she was likely struggling with usual activities more than what was initially apparent at the time of presentation. Additional contributing factors included concomitant severe anxiety, history of heavy alcohol use, systemic vascular risk, and likely OSA. In aggregate, these conditions likely reduced both her cognitive and brain reserve in managing accumulating AD pathological burden (
36). These conditions are among a host of factors associated with both development of AD (
37) and prediction of progression from MCI to AD dementia (for further details, see Table S1 in the
online supplement). Many of these factors were identified and quantified in a recent meta-analysis by Li et al. (
38). Risk factors including hypertension, cardiovascular and cerebrovascular disease, hypercholesterolemia, and history of smoking have been associated with clinical AD in other studies (
37). Although OSA was not evaluated as an AD risk factor in the meta-analysis conducted by Li et al. (
38), other studies have implicated OSA as both an important and modifiable risk factor for AD (
39). Furthermore, OSA has been significantly associated with MCI, and OSA severity is positively correlated with CSF total tau and phosphorylated tau levels, which are associated with AD pathological burden (
40).
Anxiety has been closely connected with AD in various ways. Delusions, which our patient had developed, are also common, with a 30%–35% (median) prevalence range, and they are associated with significant distress and dysfunction (
41). A significant anxiety disorder can mimic symptoms of early AD by causing executive dysfunction and resultant forgetfulness. Past or current anxiety disorder has also been implicated as a risk factor for AD, and anxiety may be directly caused by AD pathophysiology (
17,
42). Late-onset anxiety may be a prodrome to AD-related cognitive decline or occur concurrently to it, with independent neurobiological underpinnings. Evidence to date suggests that anxiety with AD is most likely associated with early neurofibrillary tangles in the entorhinal cortex, without a correlation between anxiety and cortical beta-amyloid pathology. AD pathology in the entorhinal cortex may lead to decreased sensorimotor gating of environmental stimuli, leading to a type of pathological widening of attention to potentially threatening socioemotional stimuli (
43). In addition, early neurodegeneration in the right mesial temporal lobe observed in those with AD, involving the entorhinal cortex, coupled with a relatively preserved amygdala, may mediate anxiety by allowing unregulated amygdalar hyperactivation (
43). Our patient had extensive neurofibrillary tangles in the entorhinal cortex but also prominent beta-amyloid pathology in the frontal and temporal cortices. Without serial AD biomarkers across time, it is difficult to directly attribute her worsened anxiety to pathophysiological progression of her AD or to her postmortem pathology of abundant neurofibrillary tangles in the entorhinal cortex (
43,
44). As we suggest above, her late-life anxiety exacerbation likely had a complex, multifactorial etiology, including her emotional response to daily cognitive difficulties.
The evidence for current anxiety as a risk factor for developing AD dementia is mixed, with some meta-analyses demonstrating no significant relationship (
38,
45) and others demonstrating that anxiety is a significant predictor of the progression of MCI to AD dementia (
46). The notion of anxiety and other neuropsychiatric symptoms as risk factors, or as prodromal to AD and related dementias, has been captured by the construct mild behavioral impairment (MBI) (
47). MBI is characterized by the onset of neuropsychiatric symptoms—including apathy, mood symptoms, anxiety, agitation, aggression, poor social cognition, and psychosis—that persist for at least 6 months among individuals ≥50 years of age and precede or accompany cognitive decline and dementia (
47,
48). MBI has also been independently associated with tau deposition in the hippocampus and entorhinal cortex among individuals with elevated beta-amyloid who remain cognitively intact (preclinical AD) (
49). Specifically, affective dysregulation, which includes anxiety and depression, and lack of impulse control, which includes irritability and agitation, have been shown to have direct correlation with tau pathology assessed by molecular PET and CSF assay (
49). Because the patient’s anxiety was lifelong and likely predated any early AD pathology or AD symptoms, she would not have met criteria for MBI. It is important for clinicians to assess MBI in their evaluation of patients with late-onset neuropsychiatric symptoms, especially when prior neuropsychiatric history is poorly characterized.
In summary, in our clinical evaluation of this case of late-onset AD, we used a three-tiered approach to the diagnostic formulation, as we have highlighted in all cases to date in this section of the
Journal of Neuropsychiatry and Clinical Neurosciences (
50). This involved a stepwise method of clinical syndrome identification, assessment of the severity of cognitive and functional decline, and prediction of underlying neuropathology. Our patient’s amnestic, multidomain cognitive syndrome, at the boundary between MCI and mild dementia severity, together with neuroimaging findings and cognitive testing suggested AD as the dominant underlying pathology, with likely contributions from sequelae of cerebrovascular disease, as well as anxiety and possibly heavy alcohol use and OSA. While difficult to directly attribute any changes in cognition or behavior to a particular pathological finding, the presence of mild to moderate arteriosclerosis on her neuropathological examination increases the likelihood that her systemic vascular conditions affected her brain structure and consequent cognitive decline.
Relevant to this case is that when there are confounding clinical factors, such as significant anxiety, there is often an incongruence between objectively assessed cognitive performance and patient-reported day-to-day functioning. The impact of psychosocial and environmental factors on anxiety and other neuropsychiatric symptoms is often underappreciated. The influence of these other factors underscores the importance of obtaining collateral information in evaluations and recognizing and addressing preceding or co-occurring psychiatric and medical disorders, because they often confound or mediate cognitive impairment and decline.