By all accounts, imaging genetics is a relatively new field: only the Second International Conference of Imaging Genetics was held this year. Imaging genetics combines the strengths of the very active research domains neuroimaging and molecular and statistical genetics. Although this combination has led to its sometimes being described (somewhat tongue in cheek) as a way to combine two “buzzy” fields to make them even “buzzier,” the theory behind the science is actually quite old. The basic idea is to find robust quantitative dimensional aspects of brain function or structure through neuroimaging techniques to then examine their association with specific genetic variations across individuals. Although genetic association studies have been performed for decades, the unique and revolutionary access that neuroimaging provides to structural and functional parameters of the living human brain fuels the hope of making inroads into identifying the biological mechanisms of psychiatric illness while simultaneously illuminating basic mechanisms of brain circuitry. By characterizing the impact of genetic variation on brain processes, biologically validated mechanisms emerge that can be implicated in the risk for psychiatric disorders if the genetic variant studied is associated with them. In this review I will provide a historical overview of these guiding principles and offer examples of their power and usage. To understand where our current genetic concepts come from, we must understand something about what psychiatry’s approach to genetics has been. Psychiatrists have been interested not only in what we suffer from (the diseases) but also in who we are (dimensional aspects of our cognition and personality), what we do (behaviors), and how our life events have an impact on these factors. Genetics has been applied to and in many cases has profoundly altered our perception of all of these aspects of our psychological life.
Genetics, why genes, and which ones?
Genes are the first links in a long causal chain leading from molecular biology in cellular systems to complex syndromes. Statistical genetics is a powerful technique that allows us to study the impact of genetic variation on syndromes without presupposing knowledge of the intervening biological processes. This used to be an especially attractive approach for psychiatry, in which such knowledge was critically limited; indeed, a central criticism of psychiatric disease categories is the lack of a solid basis in pathological and mechanistic understanding for the majority of our illnesses. In consideration of the fact that our current disease constructs are based mainly on criteria that are based on clinical impressions and course, it is almost ironic to consider that many of these categories were originally developed during a renaissance of medical neuroscience driven by a biological impetus: the novel science of pathological (microscopic) anatomy at the turn of the last century. Kraepelin was a contemporary of Alzheimer and Huntington, and, as for Alzheimer’s and Huntington’s diseases, he hoped to find a localizable pathological change that would provide a tangible causative link to schizophrenia, bipolar disorder, and depression.
When the hope for such a direct structural pathological change remained unfulfilled and schizophrenia, in particular, was described as the “graveyard of neuropathology” (
1), findings in statistical genetics and epidemiology provided inroads by the demonstration of sizable genetic and therefore biological contributions to many psychiatric disorders such as schizophrenia, bipolar disorder, depression, or autism. A strong indicator of the genetic contribution to a disease is heritability (
2). The early twin, adoption, and family studies were critical in this regard and provided the foundation for the rise of the statistical genetic approach that characterized the field at the turn of this century (reviewed in this issue). For schizophrenia, in a recent meta-analysis of twin studies the heritability was estimated to be 81% (
3), whereas a similar study of major depression still yielded an estimate of 37% (
4).
Given this confirmed genetic contribution, the search for the causative genes becomes a major research goal in psychiatry. It has gone through several methodological stages. The majority of initial psychiatric genetic statistical studies used linkage analysis, a technique in which genetic markers of known chromosomal position in the genome are used to identify regions on chromosomes that harbor genes for diseases (
2). Although successful for many monogenic (Mendelian) disorders, some forms of cancer (
5), Huntington’s disease (
6), and familial forms of Alzheimer’s disease (
7), linkage did not provide highly replicable findings for most psychiatric diseases (
8), suggesting that these diseases are not Mendelian in nature. In contrast to linkage analysis, genetic association methods test whether a particular genetic variant (which varies in its form; it can be a single nucleotide change, a repeated sequence, a deletion/insertion, or duplication, etc.) is enriched in affected individuals in comparison with unaffected healthy control subjects (
9). In other words, genetic association methods explore the relationship between genetic variants and phenotypic differences in the general population (
9). This direct testing of single polymorphisms is less confounded by, for example, a protective allele that could occur directly next to a causative allele, a constellation that would add “noise” to linkage analysis, does not require extended pedigrees, and has been shown to be superior in power in the setting of genetically complex disorders (
9), a key concept for psychiatric genetics in general and imaging genetics in particular.
In the past decade, with the draft of the human genome done (
10,
11) and ongoing large-scale efforts to further characterize human genetic diversity through direct sequencing of multiple individuals, we have accumulated a growing number of known gene variants (
12). Building on this base, the association approach has led to the identification of multiple genetic variants that have significant, although weak and not infrequently inconsistent, associations with psychiatric disease categories (
8). This fact has led to the realization that most common diseases with genetic components and the psychiatric disorders among them are characterized not by single major causative genes, easily identified by linkage analysis, but by multiple genetic variants in several different genes that by themselves only impart minimal risk but can in combination with each other and the environment manifest disease (
9). Conflicting results in the literature are better understood when this small amount of risk attributable to any given variant, together with the considerable variability across populations and other issues surrounding complex diseases, is appreciated (
8). Because the risk of each given variant is too small to be easily captured by linkage analysis, it is likely, and recent data indeed support this contention, that linkage regions for complex psychiatric disorders actually harbor clusters of disease genes, leading to increased linkage signals (
8,
13–
15). Therefore, of the genetic methods available, genetic association has been a particularly popular broad-based strategy for finding genes involved in mental disorders. In addition, a large number of candidate genes, whose involvement has been hypothesized a priori based on the implication of their product in a pathophysiological concept for psychiatric disorders (for example, genes encoding for neurotransmitter receptors known to be targets of psychoactive drugs or genes expressed in an anatomical structure thought to be dysfunctional), have been investigated (
8).
In addition to the genes, the application of genetics to psychiatry and imaging also requires informative dimensional target measures (phenotypes) that go beyond categorical disease or behavioral distinctions. Dimensional traits of personality and neuropsychological measures are one such phenotype. The recognition that heritable cognitive and social traits could be measured and linked to genetics is quite old. Francis Galton, Darwin’s cousin, authored an article, “Hereditary Talent and Character” (
2), in 1865 and founded the Anthropometric Laboratory in London for the measurement of “biometric” attributes and their inheritance. He plotted human behavioral and cognitive traits to dimensional curves and attempted to show their relative inheritable contribution. Galton and those who later followed in his tradition have sought to further characterize our cognitive capacities and behavioral traits through an ever-expanding list of neuropsychological scales, tests, and measurements. Although these tests are rarely believed to provide direct access to the underlying processes of the brain and are often motivated by top-down constructs of mental processes, they frequently provide robust measurements of human psychological variability that may show up to 70% heritability due to a genetic contribution (
16). Like the complex causation of the (categorical) disease phenotype, the investigation of a quantitative trait analysis is best understood in relation to the concept of multiple interacting functional common variants (
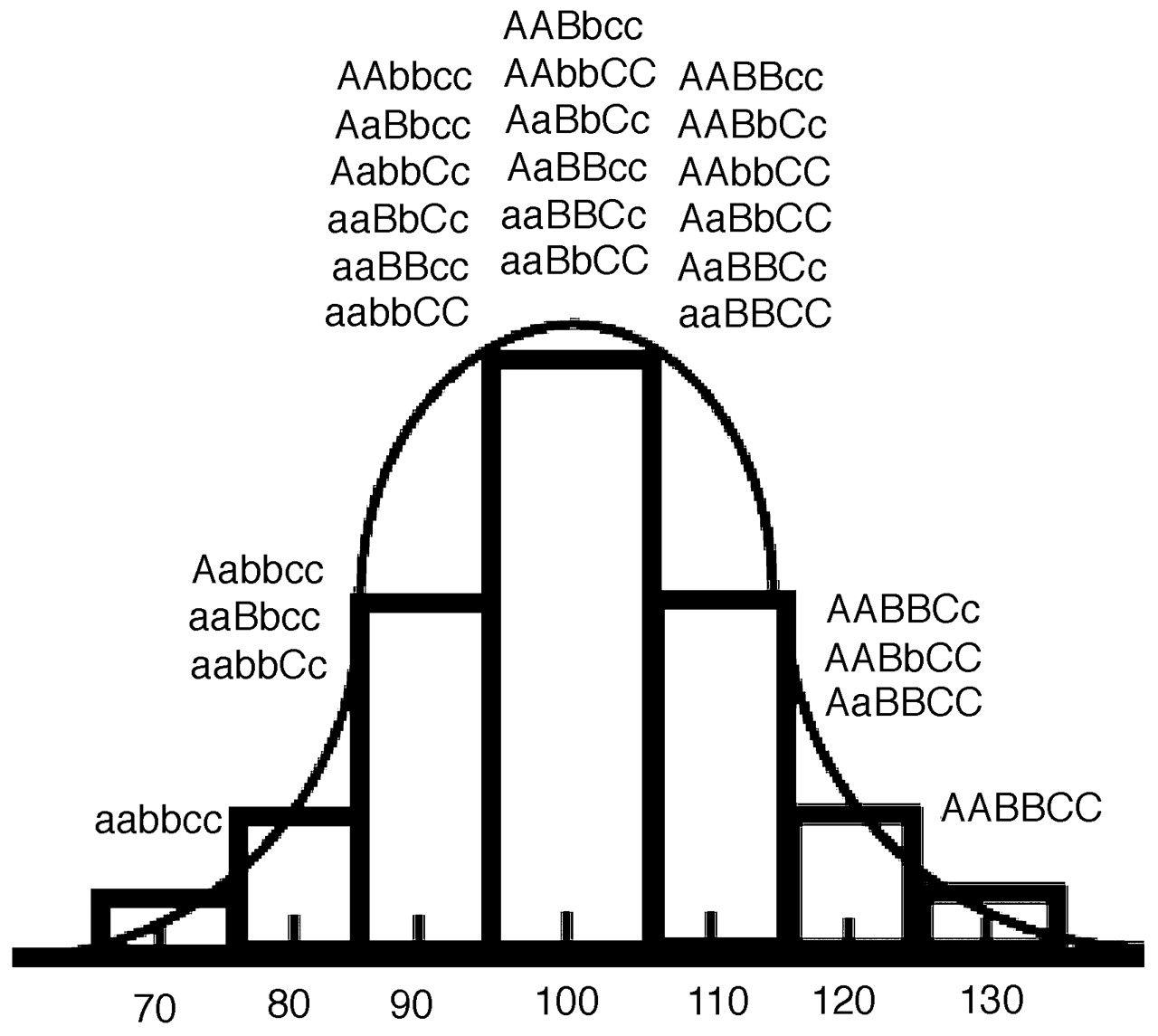
2). This theory predicts that quantitative traits should have an approximately normal (Gaussian) distribution in the population because they result from the combined contributions of many interacting genes (Figure 1).
Because the individual contribution of any single gene variant to a complex phenotypic quantitative trait is relatively minor, this theory helps us to understand the difficulty inherent in conclusively implicating a single gene in complex traits such as episodic memory or neuroticism. With complex dimensional phenotypes, placement on the curve is defined by which one of a multitude of possible combinations of variations in a group of genes an individual inherits. Usually, the number of possible combinations of these variables is greatest near the center of the distribution, whereas the extremes will be determined by a less frequent combination of all “good” or all “bad” genes (
2). Dimensional aspects of personality, cognition, and temperament have been linked with some success not only to psychiatric disorders but also to genetic variation: Examples are the association of a functional val
66met variant of BDNF with episodic memory (
17,
18), a variable number of tandem repeat polymorphisms in the promoter region of the serotonin transporter gene (5-HTTLPR; MIM# 182138) with harm avoidance or neuroticism (
19,
20), or a functional val
158met substitution in the COMT (MIM# 116790) gene with emotional reactivity (
21,
22). In these cases, as will be discussed further below, imaging genetics has been useful in identifying brain mechanisms that contribute to these observed associations.
With the use of imaging genetics, these emergent complex behavioral constructs can be broken down into their biological component parts. This adds a new dimension of understanding because quantitative personality, behavioral, and neuropsychological phenotypes reflect a single final outcome that is a product of multiple interactive processes, whereas brain imaging techniques allow the study of many individual processes closer to the level of genetic and molecular events and therefore are better suited for assessing the biological impact of genetic polymorphisms on systems-level human cognition, emotion, and behavior and susceptibility to neuropsychiatric diseases. Imaging genetics also allows the study of much smaller sample sizes than in conventional genetics because the penetrance of the genetic effects on the intermediate neural systems level is predicted to be larger than on clinical target variables; this fact is strikingly borne out by the currently available literature in which sample sizes of a few dozen subjects are often enough to show significant genetic differences (although researchers still need to remain mindful of occult confounding issues, such as inappropriate multiple comparisons or uncontrolled variation on the subject level).
The new approach of imaging genetics is fueled by a rich and ever-expanding set of genes associated with mental illness. The last meeting of the World Congress of Psychiatric Genetics provided an “official declaration” of COMT, DISC1 (MIM# 605210), G72 (DAOA; MIM# 607408), and DAT1 (MIM# 126455) as “psychiatric risk genes” (Craddock N, Rice J, Barr C, Breen G, Nothen M, Schulze T, Schumacher J, O’Donovan M, Glatt S. Summary of Gene Workshops for COMT, DAOA(G72), DISC1, and SLC6A3 (DAT): Findings. World Congress of Psychiatric Genetics, Boston, MA, Oct. 18, 2005) although associations of these genes are quite diverse and extend to a varying multitude of traits, diseases, or behavioral parameters. Overall, fully 70% of all genes are expressed in the brain and could potentially contribute significantly to the behavioral phenotypes. These associations between gene polymorphisms and behavioral, categorical, and quantitative psychiatric phenotypes provide a starting point for investigation of the underlying neural mechanisms. However, by itself genotype information does not advance our understanding of the underlying biological mechanisms, a juncture at which imaging genetics comes into play. In the next section of this review I will describe the basic methodological approaches used to characterize brain structure and function and the varied ways it has been related it to genetic variability.
Imaging
Because genes encode molecules and cannot directly cause complex phenotypes such as hallucinations or complex behaviors such as alcoholism, it is necessary to examine the effect genetic variation has on the organ system that generates the complex behaviors, the brain. Multimodal imaging approaches can identify genetic influences on brain structure, function, and pathology. With modern methods of magnetic resonance imaging (MRI), functional MRI (fMRI), positron emission tomography (PET), single photon emission computed tomography, and EEG/magnetoelectroencephalography we can noninvasively and often repeatedly collect measurements on brain function and structure. These imaging techniques provide access to a systems-level description of the relevant neurobiology that allows for relating the underlying cellular and genetic processes to the cognitive and psycho-pathological domain.
The structure of the brain is readily visualized using modern magnetic resonance techniques and has been for decades. The main research problem with structural imaging psychiatry is the need for a reliable objective methodology to measure the subtle structural differences associated with psychiatric disorders in general and genetic variation in particular. Supplementing and extending early work which included measurements of cerebral volumes (usually manually and laboriously delineated) of cortical structures defined by macroscopic anatomical features (for example, prefrontal cortex or amygdala volumes), a method that is not constrained by such subjective and shifting landmarks has been widely applied in imaging genetics: voxel-based morphometry (VBM) (
23). In this sophisticated whole brain technique, local changes in grey matter volume are mapped simultaneously and analyzed without being constrained by conventional landmarks, a considerable advantage when one is studying genetic mechanisms for which changes often do not follow features defined by macroscopic anatomy. This technique is also largely automated and ideal for examining the large sample sizes required for genetic studies.
Further structural insights can be expected from diffusion tensor imaging (DTI) (
24). DTI is a magnetic resonance approach that permits mapping of the direction and integrity of white matter tracts, which provides important information about regional connectivity. This method has yet to be applied to genetic questions in psychiatry but may help elucidate questions of connectivity and the actions of acting genes that have an impact on white matter. Another set of MRI techniques, magnetic resonance spectroscopy (MRS), allows the mapping of aspects of brain metabolism (
24). Whereas structural MRI is sensitive to hydrogen signals, MRS can characterize complete resonance spectra and can be used to measure more complex molecules, such as
N-acetylaspartate, a putative marker of neuronal integrity, and in the near future also neurotransmitters such as glutamate and γ-aminobutyric acid (GABA). This technique has already been applied to studying the genetic impact on neuronal integrity and will probably be expanded to probe biochemical changes linked to genotype.
With use of the same magnetic resonance technology, since the early 1990s, physiological alterations of brain function have been investigated with fMRI (
25). This technique is used to measure regional increases in relative blood flow that are correlated to increased neuronal activity. In the beginning, fMRI experimental procedures were rather simple, usually employing a contrast in blockwise alternation of different stimulation conditions. Since then, the methodological spectrum has expanded to event-related task designs, which allow more sophisticated analysis of brain responses to brief stimuli under conditions that can rapidly change. A relatively recent advance in the analysis of functional imaging is the characterization of functional connectivity between brain regions, which allows for the characterization of aspects of network interactions in human brain (
26). Examples of the application of these techniques to imaging genetics will be given later. Further technical developments, including those in computational power and data storage, led to the development of MRI scanners with ultra-fast gradient systems. Today, multichannel radiofrequency coils (array coils) can decrease acquisition time and/or increase signal-to-noise ratio and spatial resolution substantially by simultaneous measurement of partial volumes. By using these advanced acquisition schemes, whole brain data collection with highly resolved slices is now routinely done within a few seconds. These advances in increasing the spatial and temporal resolution and image quality and the ability to be used noninvasively and repetitively administered on most standard scanners, combined with more complex cognitive paradigms, have made fMRI the current functional method of choice for imaging genetics (
25).
Functional imaging genetics has not been limited to fMRI; PET imaging is also used (
24). PET is a powerful and versatile multimodal technique predicated on the use of radiolabeled injected markers emitting positrons that annihilate with electrons, sending out pairs of photons of known energy that can be detected with high spatial resolution. Depending on the labeled molecule, PET can be used to study neural activation through mapping blood flow with labeled water, energy metabolism through [
18F]fluorodeoxyglucose, a marked glucose analog, or multiple aspects for biochemical kinetics such as neurotransmitter turnover, transporter or enzyme activity, or accumulation of metabolic products. However, the use of this technique has been somewhat limited by its greater expense, the need for a dedicated radiochemical facility for many tracers labeled with short-acting isotopes, and radiation safety limits, which restrict the amount of exposure and make longitudinal studies impractical.
Imaging methods have been used for more than a decade to identify functional differences between disease groups, for example, patients with schizophrenia versus control subjects (
24), or between cognitive states, for example, comparing a cognitive task such as looking at emotional faces with a control task, looking at scrambled images (
24). Because of the inherent variability in both the target measures and the underlying putative neural substrate, groups of people and/or repetitive measures of events are usually needed to find meaningful differences. These studies have considerably advanced our knowledge of brain circuits associated with the performance of specific cognitive tasks and also pointed toward some systems differentially active in groups of psychiatric patients relative to control subjects, such as prefrontal or hippocampal function and their interactions in schizophrenia (
26) or activation of the limbic system in patients with affective or anxiety disorders (
27). The unique power of neuroimaging to characterize brain structure and function has led to considerable refinement and extension of the concept of a phenotype. From the point of view of genetics developed above, neuro-imaging measurements provide a large number of physiological quantitative traits, for example, the relative changes in blood flow in several regions of the brain or changes in gray matter volumes.
An instructive example of defining the phenotype is afforded by dopaminergic modulation of prefrontal function, which is critical in interactions with midbrain and striatum for motivated behavior, working memory (
28), and reward-related learning (
29). A compelling application of imaging genetics involves prefrontal function in conjunction with a variation in the gene encoding catechol-
O-methyltransferase (COMT), a major enzyme degrading cortical dopamine. Dopamine action at the synapse is terminated either by reuptake by the do-pamine transporter or via diffusion out of the synapse or catabolism by COMT. Because dopamine transporters are scarce in prefrontal cortex (
30), COMT is a critical determinant of prefrontal do-pamine flux, as confirmed by mouse knockout models (
31) and in vivo microdialysis (
31). The gene is located at 22q11.2, a region implicated in schizophrenia by linkage (
32) and by the 22q11.2 deletion syndrome (MIM# 192430), a hemideletion associated with a 30% increased risk of schizophrenia-like illness (
33). A single nucleotide change leads to a common val
108/158met amino acid substitution that affects the stability of the COMT protein and leads to a significant decrease in the activity of the enzyme in brain and lymphocytes (
34). In one of the first applications of neuroimaging genetics, Egan et al. (
35) used a reliable activator for prefrontal cortex, the N-back working memory task, to demonstrate that this coding variant exerts an effect on prefrontal cortex activation. In agreement with this result, a variation in COMT was also found to modulate prefrontal cortex-dependent neuropsychological performance (
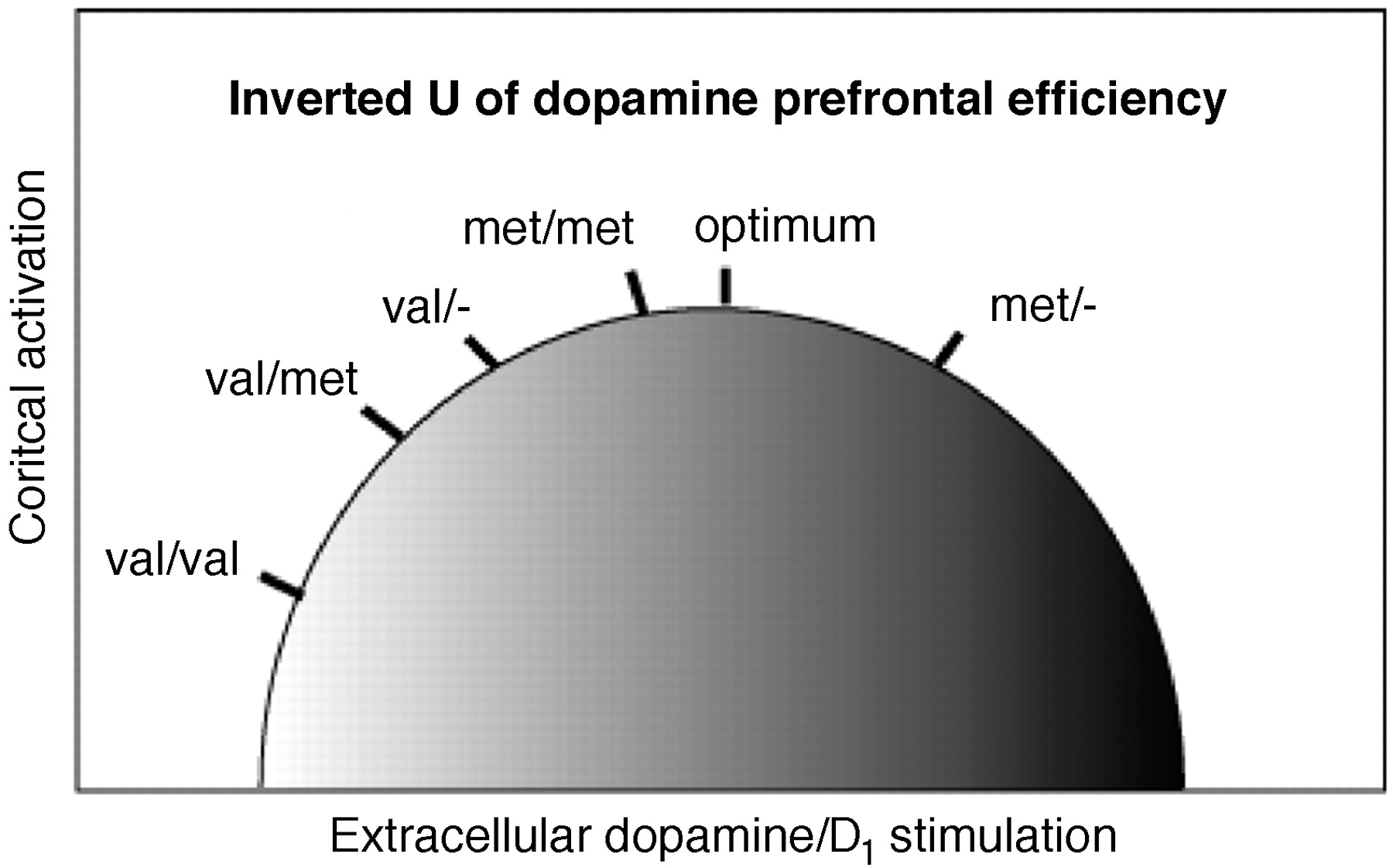
18) Specifically for prefrontal activation during working memory, an “inverted U–shaped” relationship between working-memory related activation of prefrontal cortical neurons and dopaminergic, especially D
1 receptor, stimulation has been established in a large body of work (
28,
36,
37), with dopaminergic tone essential for optimizing the signal-to-noise ratio or tuning in prefrontal networks (Figure 2). That placement on this curve depends on the
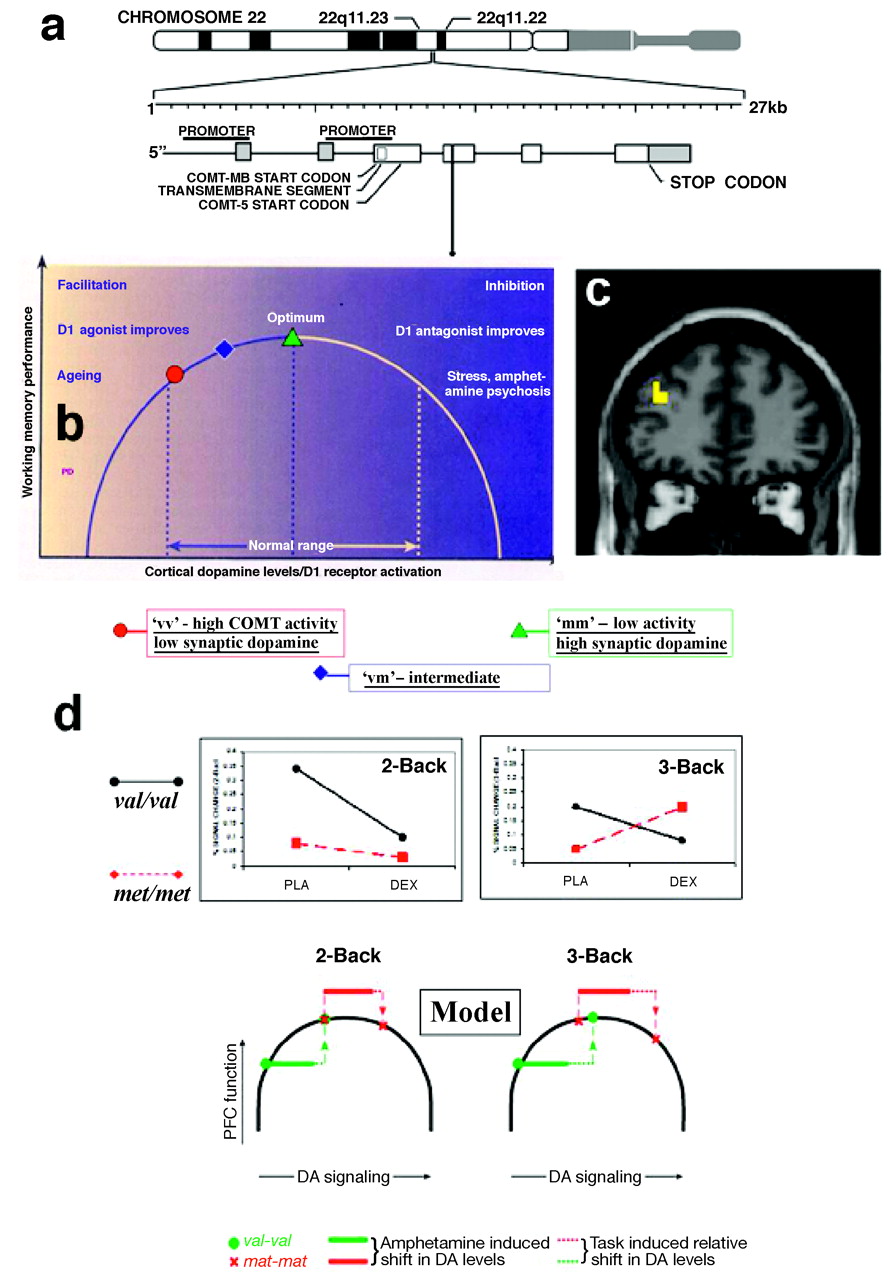
COMT genotype was demonstrated in an elegant study by Mattay et al. (
38), combining imaging genetics with a drug challenge, in this case with the dopaminergic agent amphetamine, to characterize this complex effect. The authors showed that during performance of a demanding working memory task, homozygous
val carriers increased the efficiency of their frontal lobe response under amphetamine, whereas both the efficiency of prefrontal activation and the performance deteriorated in subjects homozygous for the
met allele who were given the drug, suggesting compromised information processing (Figure 3). This finding indicated that amphetamine “pushed” dopamine levels in these individuals beyond the optimal range on the inverted U via activation of inhibitory mechanisms, such as inactivation of N-type Ca
2−channels (
39), activation of GABA-ergic interneurons (
40), and pre- and postsynaptic reduction of glutamate-mediated synaptic responses (
41) (Figure 2). Thus, a measurable intermediate phenotype brain response that may be modifiable and could be directly and longitudinally assayed in therapeutic research is provided. Additional evidence for the tuning concept came from a PET study (
42) showing that the COMT genotype affects prefrontal regulation of midbrain dopamine synthesis in a genotype-dependent directionality consistent with the inverted U–shaped model (
42,
43). Increased noise in
val carriers provides a plausible mechanism underlying the (albeit weak) association of this allele with schizophrenia, although meta-analytic studies suggest that this effect is only barely penetrant on the level of diagnosis (
44) and is therefore likely to be modified by other genetic variants, possibly even within the same gene.
An intriguing experiment of nature giving independent evidence both of the inverted U model as such and of the implication of genetically mediated prefrontal inefficiency in schizophrenia risk is provided by the 22q11.2 hemideletion syndrome, for which recent data show that
met allele carriers are at higher risk for psychosis and structural brain change (
45). This at first view counterintuitive result is readily accommodated by the pathophysio-logical model. Because one copy of the COMT gene is missing in this syndrome, COMT activity is already considerably reduced. Therefore,
met allele carriers will now be suboptimal because they are positioned to the right of the optimum of the inverted U–shaped curve, whereas
val carriers are closer to optimal. Illustrating the concept of risk alleles of minor isolated contribution, Egan et al. (
35) calculated that COMT only accounted for 3% of the variance even on the directly affected intermediate brain phenotype, prefrontal cortical activity in the N-back task.
The concept of multiple interacting variants de-fining a quantitative trait also predicts that other genetic risk alleles should have an impact on pre-frontal function. This is indeed what was found for GRM3, encoding a metabotropic glutamate receptor modulating synaptic glutamate (MIM# 601115), that has been proposed as a candidate gene linked to schizophrenia (
46) and therefore potentially affecting prefrontal activity. With use of the same prefrontal activating task (the N-back), it was shown that a single nucleotide polymorphism in GRM3 predicted prefrontal activation and additionally
N-acetylaspartate, an in vivo MRS measure related to synaptic activity closely correlated with tissue glutamate (
46). This group further went on to study the effects of G72 (an activator of D-amino acid oxidase), which is involved in the metabolic pathway of D-serine, an agonist of the
N-methyl-D-aspartate receptor glycine site) on working memory (
47). While characterizing other genes, BDNF (brain-derived neutrotropic factor), DISC1 (a structural protein highly expressed in the hippocampus), and G72, implicated in both bipolar and/or schizophrenia, they also implicated these genes in hippocampal dysfunction and neurocognitive variables of working and episodic memory (
17,
18,
47,
48). Other genes will surely follow, and it will then be of interest to characterize them and the interactions between them using imaging genetics methodology. This characterization will give us direct measures of whether our risk genes are additive or whether their combined effects are more complex.
Another interesting approach does not consider prefrontal activation as such but rather interactions between prefrontal cortex and other structures. For example, functional circuits linking prefrontal cortex and striatum (
49) and hippocampus (
26) have been implicated in schizophrenia, and early results are already being seen with the recent study of the impact of G72 on all these functions (
47). These regions have been individually shown to be modulated by variations in risk genes, as have been the prefrontal-midbrain interactions relevant for regulated dopamine release reviewed above, but an imaging genetic characterization of the circuit has yet to be done. As other pathological processes are revealed through imaging, such as an increased rate of cortical thinning in childhood-onset schizophrenia (
50), we increase our repertoire to identify genetic contributors to these new phenotypes (
51) that will advance work toward discovering molecular mechanisms.
Turning to another disorder, one of the first success stories both in the identification of risk alleles for genetically complex psychiatric disorders and for the application of imaging genetics was the identification of mechanisms linking early age of onset of Alzheimer’s disease with the APOE4 risk allele (MIM# 107741). Since then, results from imaging genetics have informed the prognosis, pathology, and mechanistic underpinning of Alzheimer’s disease. APOE is involved in cell maintenance and repair, including amyloid clearance. Functional brain imaging studies have consistently shown APOE4 effects even in clinically normal individuals. Early resting studies, using PET, showed widespread reductions in glucose metabolism in otherwise normal APOE4 carriers (
52,
53) that were interpreted as presymptomatic cerebral impairment. Functional circuits involved in episodic memory were specifically implicated when Smith et al. (
54) used fMRI to demonstrate reduced activation in APOE4 carriers in medial temporal lobe during fluency and object recognition. During an episodic memory task in asymptomatic subjects who preformed the task at the same level, Bookheimer et al. (
53) found the opposite result, increased activity, in APOE4 carriers, possibly reflecting a compensatory response: APOE4 carriers had to recruit more neuronal activity to achieve the same behavioral result. This latter finding was independently replicated in other regions (
55,
56), including the prefrontal cortex (
55). A recent large study confirmed a correlation between gene dosage and abnormal glucose metabolism in regions of the precuneus, posterior cingulate, and parietotemporal as well as frontal cortex (
57). The earlier study by Bookheimer et al. (
53) also indicated a prognostic dimension to the imaging genetic findings, because these authors found that baseline brain activation correlated with subsequent verbal memory decline. Recent results using PET suggest that abnormalities may be discernible in at-risk individuals as early as in the age range of 20 to 40 (
84).
Multimodal genetic imaging has been used to further characterize the underlying mechanisms in Alzheimer’s disease. Because the cholinergic system has been implicated in the genesis of memory impairments in Alzheimer’s disease, Cohen et al. (
58) used the
18F-labeled muscarinic-2 selective agonist 3-(3-(3-[
18F]fluoropropyl)thio)-1,2,5-thiadiazol-4-yl)-1,2,5,6-tetrahydro-1-methylpyridine to directly measure the muscarinic component of the cholinergic system as a function of the APOE4 genotype with PET. Their results suggested that APOE4 carriers have increases in the number of unoccupied muscarinic-2 receptors, which could reflect lower synaptic acetylcholine. This technique will probably also be applied in Alzheimer’s disease. With appropriate methodology, imaging genetics can show not only neurochemical but also patho-anatomical features of disease as a function of genotype, for example, by new ligands that can be used for radiolabeling of amyloid proteins and in vivo mapping of amyloid plaque density and neu-rofibrillary tangles in the human brain (
56,
59).
Imaging genetics has also been used to help parse out genetic contributions to dimensional traits and mechanisms for susceptibility to disease in gene–environment interactions. An instructive example is afforded by the study of how trait analysis and neuroimaging were combined to uncover the possible mechanisms of affective disorder susceptibility. The connection between anxiety and major depression has been demonstrated through twin and family studies (
60), indicating that this personality trait, for example, measured by the harm avoidance scale of the commonly used triphasic personality questionnaire, is linked to risk for depression and anxiety. Considerable evidence, not in the least from the efficacy of serotonin reuptake inhibitors and other drugs modifying synaptic serotonin, implicates the serotonergic system in anxiety and depression. It was therefore a finding of great interest when Lesch et al. (
61) demonstrated a genetic effect on trait anxiety linked to this neurotransmitter system. They studied a common variable number tandem repeat polymorphism in the 5′ promoter region of the serotonin transporter SLC64A (5-HTTLPR), where the so-called short (
s) allele resulted in a reduction of transcription and was associated with increased trait anxiety. During intense further study of this common polymorphism, weak and inconsistent evidence was uncovered for an association of the
s variant with risk for depression. A landmark result by Caspi et al. (
62) clarified a major reason for the limited power of a purely genetic association study in this setting. In a large longitudinal study of a cohort from New Zealand, these authors showed that the
s allele indeed predicted risk for depression, but only in interaction with environmental adversity; without averse experience, the main effect of genotype was minimal. This intriguing result was widely received as being paradigmatic of the complex risk structure of psychiatric disorders, but the neural mechanisms mediating this complex association were unclear. An important first step in elucidating these mechanisms was taken in an imaging genetics study by Hariri et al. (
63). Using fMRI, these authors explored the effect of the 5-HTTLPR polymorphism on the activation of the amygdala, a key structure in the signaling of environmental danger that has been strongly implicated in emotional processing and anxiety. Using a task that produced robust and reliable activation of the amygdala, Harari et al. (
27) showed that subjects carrying the
s allele had a significant increase in their amygdala activity. These results have now been replicated by several groups in several different populations. Recent multimodal neuroimaging studies have aimed at further dissecting the neural circuits mediating increased amygdala activation in this setting. Pezawas et al. (
64) used VBM to show that
s carriers had reduced gray matter volume not only in the amygdala, but prominently in the perigenual anterior cingulate cortex. This result was biologically intriguing as the subgenual cingulate displays the highest density of 5-HTT terminals within the human cortex (
65) and is affected by antidepressant drugs selectively affecting the serotonin transporter (
66 –
68). Animal models demonstrate that even transient alterations in 5-hydroxytryptamine homeostasis during early development modify the neural connections implicated in mood disorders and cause permanent elevations in anxiety-related behaviors during adulthood (
69 –
71). This finding suggested that abnormal wiring of this circuit in the
s allele carriers, a hypothesis that was further explored by using fMRI to characterize functional interactions between the amygdala and cingulate (
64). The results differentiated two areas of the cingulate cortex: one, the subgenual cingulate, was positively coupled with amygdala and the other, the supragenual cingulate, was negatively coupled. This imaging result reflected a known anatomical feedback circuit in nonhuman primates from the amygdala to the rostral cingulate to the dorsal cingulate and back to the amygdala. Importantly, convergent evidence strongly suggests that these amygdalacingulate interactions represent a functional feedback circuitry that regulates amygdala processing of environmental adversity. Stimulation of perilimbic prefrontal cortex inhibits amygdala function (
72), and lesions of this region markedly impair fear extinction (
73). Genotype had a strong impact on wiring in this circuit: The 5HTTLPR
s allele carriers had a functional disruption in showing reduced connectivity of the subgenual anterior cingulate cortex and the amygdala, suggesting a reduction of feedback inhibition (
65). This reduced feedback inhibition could therefore underlie previously observed increased activation of the amygdala and the reduced inhibition when these carriers were presented with emotional stimuli. Most importantly, the likely function of this circuit in fear extinction provides a plausible neural substrate for the impact of early adversity, which would probably produce the kind of fearful associations that require a functional extinction mechanism to erase. Given the reviewed evidence for an impact of serotonergic overstimulation on neuronal maturation, the proximate mechanism was felt to probably be abnormal neurodevelopment of this system as characterized by the VBM structural images. An important test of these concepts was provided by relating them back to the level of trait behavior. Although previous attempts to correlate amygdala reactivity or single structural measures with behavioral scores of harm avoidance or neuroticism were unsuccessful, Peza-was et al. (
64) found that 30% of the variance of behavioral scores of harm avoidance was captured by the amygdalasubgenual anterior cingulate cortex functional connectivity, indicating that imaging characterization of network properties may be of crucial importance to identify neural mechanisms mediating genetic effects. These studies therefore provide a fuller account of the mechanisms underlying trait anxiety in terms of the disruptions of a functional inhibitory circuit and consequent decreased inhibition of the amygdala, leading to a hyperresponsive emotional response in the setting of prominent life stressors, which contributes to an increased risk for major depression.
A further example of how imaging genetics can be used to parse mechanisms of complex behaviors disorders and dissect them at the level of brain mechanisms is recent work on genetic contributions to violence. Within the field of violence research, a distinction is commonly made between impulsive aggression on the one hand and instrumental aggression, linked to psychopathy and antisocial personality disorder on the other. Although this distinction is intuitively compelling, both of these factors are highly correlated in populations of violent offenders and contribute to high scores on the psychopathy checklist (
74), and mechanistic means to separate them were lacking. A genetic approach to disentangling these components of violence on the neural level was provided by a recent study of variation in the X-linked gene encoding monoamine oxidase A (MAOA; MIM# 309850). MAO is a key enzyme in the catabolism of mono-amines, especially serotonin, and provides the major enzymatic degradation of norepinephrine and serotonin during brain development. In mice, knocking out this gene results in abnormal emotional learning (
75) and increased aggression in males (
76). In a Dutch kindred with an inactiving missense mutation in MAOA, males carrying this inactive gene exhibited violent criminal behavior (
77). Whereas such null variants are very rare, a common functional variable number tandem repeat polymorphism in this gene,
MAOA-L, has an impact on the level of expression and had previously been associated with violent offenses in males with maltreatment in childhood (
78 –
81). By using a combination of functional imaging tasks to study response inhibition in the flanker task, emotional regulation in an emotional faces matching task, a covert emotional encoding and retrieval task, as well as structural neuroimaging, neural circuits affected by this gene variant were examined in a recent imaging study (
82). In a population of normal control subjects,
MAOA-L predicted relative decreases in the volume of the limbic system and hyperresponsivness of the amygdala. The authors also saw diminished reactivity of regulatory prefrontal areas. When the men, who only have a single copy of the gene, were separated out, the
MAOA-L allele also predicted increases in the volume of the orbito-frontal volume, as well as amygdala and hippocampus activation during an emotional memory task. Finally, and again in men only, a regulatory region in prefrontal cortex showed reduced activity during response inhibition. The orbitofrontal and cingulate cortex and interactions of these prefrontal regions with amygdala have been implicated in fear extinction and reinforced learning, which may partly underlie the observed interaction of the genetic effect with early adverse experience, which is expected to tax these neural circuits. In addition, by showing hyperreactivity of the amygdala together with reduced activity of areas responsible for cognitive inhibition, these neural circuits are expected to contribute to the risk for impulsive, but not instrumental, aggression suggest that these dimensions of complex behavior may be dissociable by an imaging genetic strategy.