After many years of painstaking work, efforts to identify susceptibility genes for schizophrenia have yielded several long-awaited results since August, 2002. Dystrobrevin-1 binding protein (dysbindin), neuregulin-1, catechol-
O-methyltransferase (COMT), G72,
d-aminoacid oxidase (DAAO), regulator of G-protein signalling-4 (RGS4), and Proline d-hydrogenase (PRODH2) have been identified as susceptibility genes in one, and in most cases more than one sample (reviewed by Harrison and Owen (
1). Despite this success, complex traits remain daunting phenotypes for the geneticist to study. Incomplete penetrance, epistasis, pleiotropy, imprinting, genetic heterogeneity, and phenocopies conspire to make the pathological entity in question a moving target.
It has become clearer in recent years that the genetic complexity of schizophrenia as produced by the above mechanisms is compounded by a second, related level of complexity. This additional level is the influence of genetic factors on phenotypic variation. It has long been noted that there is substantial variation in the presentation of schizophrenia. Individual patients may differ greatly with respect to age of onset, course of illness, premorbid psychopathology, interepisode recovery, and the prominence of a range of clinical features such as hallucinations, delusions, thought disorder, and negative symptoms. Indeed, experienced clinicians often remark that it is quite possible to encounter patients with no features in common. Taxonomic schemes have long attempted to account for such clinical heterogeneity even before Kraepelin unified earlier conceptions such as
paranoia,
hebephrenia, and
catatonia under the rubric
dementia praecox (
2). These categories were subsequently operationalized in DSM and ICD subtypes (
3). Later, psychopathological features were reduced to several dimensions using factor analysis, the most prominent of which have been the positive, negative, and disorganization symptom dimensions (reviewed by Peralta and Cuesta (
4).
The last few decades have seen an accumulation of data strongly suggesting that familial factors can influence not only the liability to schizophrenia but also the specific clinical character of the illness. These data in turn raise several questions. Among these are (a) whether the different clinical forms of illness are produced by distinct familial/genetic mechanisms, and if so, (b) whether these mechanisms act only once individuals are affected, and (c) whether they also affect personality or other traits in nonpsychotic relatives. These questions are of central importance in efforts to increase the power of linkage studies to detect susceptibility genes. Answers to them, for example, could be instrumental in developing more genetically homogenous phenotypes to increase the signal to noise ratio in linkage and association studies, as well as increasing statistical power by including subjects who inherit susceptibility alleles, yet do not meet illness criteria. Clarifying the relationship between genotype and phenotype, as well as developing conceptual and statistical means of increasing the power to detect linkage and association, will be vital in facilitating further gene-mapping efforts in psychiatric illnesses.
The purpose of this article is to propose a framework for models of gene action in psychiatric illness that attempts to account for a seemingly disparate body of findings, and to explore the impact of these models on the design and interpretation of genetic linkage and association studies. We hope to stimulate discussion and further scientific work in an area that is currently lacking in theoretical underpinnings, yet may have significant implications for current and future gene-finding studies as well as our understanding of the genetic architecture of psychiatric illness and other complex diseases. We have chosen schizophrenia as the illness with which to illustrate these concepts, but they may be extrapolated to other psychiatric as well as nonpsychiatric complex traits.
Clinical and genetic heterogeneity
It is by now widely accepted that the clinical presentation of schizophrenia is variable and multidimensional. However, the cause of this variability remains unknown. The most intuitively appealing explanation is that clinical heterogeneity is due to heterogeneity in underlying etiology. Genetic heterogeneity has long been suspected, and is even more strongly supported now given the rapid developments in the molecular genetics of the illness.
The first efforts to test the hypothesis that clinical heterogeneity was due to etiological heterogeneity were family studies. These tested whether specific clinical subtypes of schizophrenia differed with respect to the risk of illness in relatives, mostly reporting that probands with high levels of negative symptoms were more likely to have relatives affected with schizophrenia (
5–
8). This suggested that these cases had a particularly high familial or genetic liability to illness. However, not all studies have found this relationship (
9). A second approach was to examine the clinical resemblance of family members that are affected with schizophrenia, who are likely to inherit the same susceptibility alleles. Several studies have reported that affected sib-pairs are correlated for clinical features such as age of onset and symptomatic profile (
10–
12).
These two approaches suggest that familial factors affect not only the general liability to illness but also influence the clinical form of the illness. However, these studies cannot truly examine the relationship between symptoms and genetic risk, as shared environment could theoretically cause the same associations. A recent meta-analysis of twin studies of schizophrenia has confirmed that shared environment probably plays a significant if small role in the etiology of schizophrenia (
13). Studies of psychotic twin pairs, although few in number and with small sample sizes, have reported significantly greater resemblance for some clinical features in monozygotic than dizygotic twins, suggesting that familial factors affecting these features are in fact genetic (
14,
15).
A much more direct approach is to test whether specific susceptibility genes are related to more or less specific illness presentations. One of the earliest efforts to relate clinical and genetic heterogeneity
per se was performed in the Irish Study of High Density Schizophrenia Families (ISHDSF). Family LOD scores at linked regions on chromosomes 5q, 6p, 8p, and 10p were regressed onto their affected members’ lifetime symptom scores from the Major Symptoms of Schizophrenia Scale (
16). On chromosome 8p, positive LOD scores were related to several symptoms resembling the “core” schizophrenia phenotype, such as affective deterioration, thought disorder, poor outcome, and low levels of depression. In a related study, in a sample of 65 sibling pairs from Maryland, pairs who shared two alleles at a chromosome 8p marker and one allele at a chromosome 14 marker were more likely to have bizarre delusions, attendance of a special school, affective symptoms early in the course of illness, and seizures (
17). As several susceptibility genes have recently been identified and replicated, it is now possible to examine the relationship between specific risk alleles and the clinical phenotype. Our group has recently found that high-risk alleles in dysbindin are preferentially transmitted to patients with high levels of negative symptoms (Fanous
et al., unpublished results), while the val allele in COMT is preferentially transmitted to patients with high levels of affective symptoms (McClay
et al., unpublished results). These studies suggest that certain susceptibility alleles or combinations thereof confer liability to more or less specific forms of psychotic illness.
Susceptibility vs modifier genes
However, several lines of evidence also suggest a discontinuity between clinical features and susceptibility genes. In the ISHDSF, linkage to chromosomes 5q, 6p, and 10p had no impact on any of the clinical features examined. The significant sibling correlations for clinical features found in this sample are therefore more likely to be due to familial factors other than the susceptibility genes in these regions. Some clinical features do not predict risk in relatives, also consistent with this hypothesis.
A number of association studies have been published demonstrating that clinical features were associated with specific polymorphisms
within a sample of psychotic subjects. These have reported associations between polymorphisms in DRD4 and catatonia (
18) and delusions (
19), DRD2 and disorganization and delusions(
20) CCK and positive symptoms (
21), 5-HT promoter and auditory hallucinations (
22) and affective symptoms (
23), HkCa3 and negative symptoms (
24) and BDNF and DAT1 and negative symptoms (
23). Moreover, these polymorphisms were not associated with illness in these samples.
These studies suggest that a number of genes may influence clinical features associated with the illness without altering the liability to illness itself, that is, are modifier genes. We define modifier genes as genes that influence disease characteristics, but that do not, themselves, alter disease liability. It would be plausible that a number of modifier genes influence the clinical form of psychotic illness, as they have been demonstrated to affect the clinical expression of a number of other diseases, including hereditary deafness (
25), cystic fibrosis (
26), and Parkinson’s disease (
27). In the first genome-wide linkage study aiming to detect modifier loci for clinical features of schizophrenia, Cardno
et al. (
28) reported suggestive linkage on chromosome 17q to age of onset in a sample of 77 concordant sibling pairs. Our group has found suggestive evidence of linkage of the course of illness to two chromosomal regions that are not linked to schizophrenia (Fanous
et al., unpublished results).
As a caveat, it has long been noted that levels of symptoms of psychotic illness change to varying degrees with time (
29,
30), for example, positive symptoms tend to diminish with increasing duration of illness, while negative symptoms are either stable or increase. Genetic studies of psychiatric illness examining either clinical features or clinically defined groups of subjects are therefore both susceptible to measurement error resulting from the temporal instability of clinical features and/or medication effects. This could be addressed at least in part by using lifetime rather than cross-sectional assessments of psychopathology.
The effects of genes in nonpsychotic relatives
While accumulating evidence suggests that the clinical features of schizophrenia are affected by both susceptibility and modifier genes, it has been unclear what, if any, are the effects of these on non-psychotic relatives. Since the time of Kraepelin, the nonpsychotic relatives of schizophrenics have often been noted to manifest quantitative abnormalities in personality (
31,
32). These findings were later extended to other “endophenotypes,” such as cognition (
33,
34), psychophysiology (
35,
36), and brain structure (
37,
38) that are thought to be related to the expression of genes relevant to the etiology of schizophrenia (reviewed by Siever and Davis (
39).
This suggests that disease states and normal traits may lie on opposite ends of a continuum of liability. An implication of this is that susceptibility alleles for schizophrenia may not confer risk in an “all or none” fashion, but rather, may be quantitative trait loci (QTLs) whose effects are variable depending on genetic background and environmental factors. Empirical studies have begun to support this. We have recently found that LOD scores of a genome scan of narrowly defined schizophrenia correlated with LOD scores of schizotypal traits in unaffected relatives (Fanous
et al., unpublished data). The Val158Met polymorphism in catechol-
O-methyl transferase is the only putative susceptibility allele studied to date in unaffected individuals. In a Greek sample, the Val allele had a higher frequency in individuals with high levels of self-reported schizotypy (
40). The same allele was associated with decreased performance on several cognitive tasks in control subjects, in which schizophrenics also perform poorly. These include the
n-back task, a test of working memory and attention (
41), as well as the Wisconsin Card Sort, a test of executive function (
42,
43).
A remaining question has been whether the effects of some susceptibility or modifier genes on clinical features are observable in nonpsychotic individuals. In the Roscommon family study, negative symptoms in psychotic probands predicted negative symptomlike schizotypal features such as social isolation in nonpsychotic relatives. Furthermore, positive symptoms in psychotic probands predicted positive symptom-like schizotypal symptoms, such as magical thinking and illusions, in nonpsychotic relatives (
31). This suggested that the familial factors influencing the clinical features of schizophrenia have effects in the same direction on related personality traits in nonpsychotic individuals.
Further progress in understanding the effects of high-risk genotypes on phenotypes in nonpsychotic individuals could benefit from advancements in measuring these phenotypes themselves. First of all, the reliability of these measures has been studied far less than that of the diagnosis of severe mental illnesses such as schizophrenia. Furthermore, there are more commonly used assessment instruments for phenotypes such as schizotypy as compared to diagnostic systems for schizophrenia, making the interpretation and contextualization of results more difficult. This may be in part because of their attenuated or subclinical nature, which shades gradually into normal states, and because they less frequently come to clinical attention. More thorough testing of the reliability of such measures and possibly the development of newer ones will therefore be required. A confound that may be more difficult to overcome is the transient effect of emotional and other states on trait measurements of personality. This has been more thoroughly studied in traits representing negative emotionality, such as Neuroticism, in which depressive states are clearly associated with higher concurrently measured levels of the trait (
44,
45).
A taxonomy of gene action
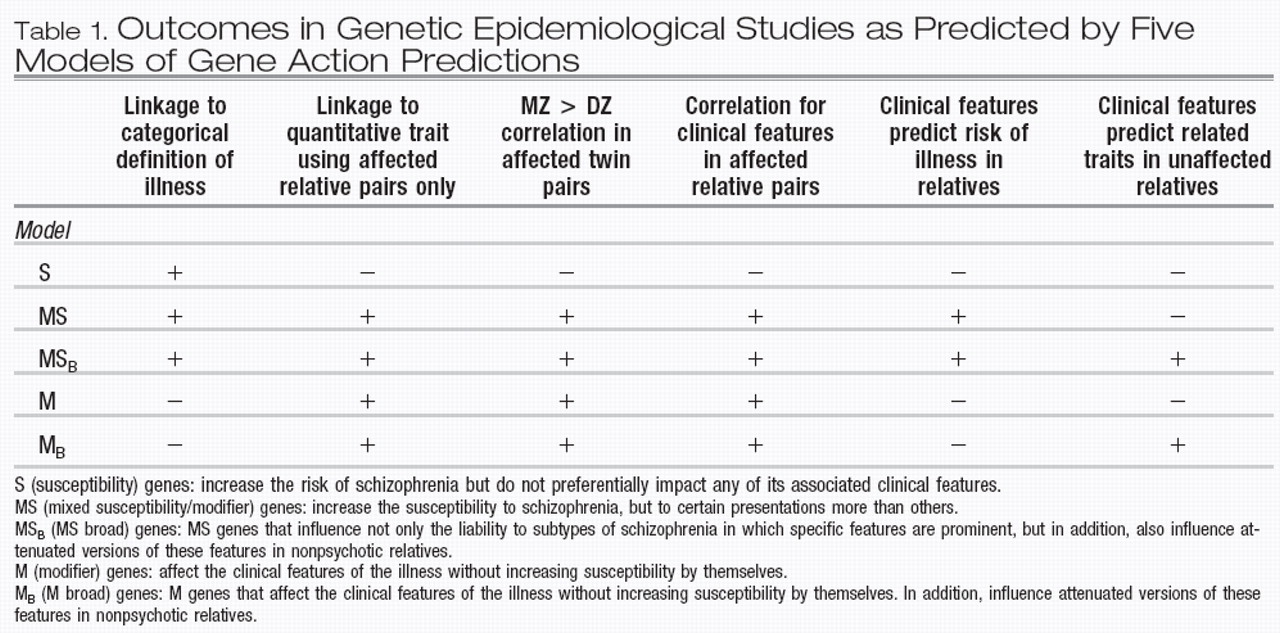
In examining the data on the impact of familial/ genetic factors on clinical variation in schizophrenia, we could discern five specific ways in which genes may impact clinical features in psychotic pro-bands and their nonpsychotic relatives. In this section, we describe five types of “genes” and their expected effects on the outcome of family and twin studies, the results of which could provide a way to distinguish between them. It is clear that these considerations are only heuristic, however, as any instantiation of illness could come about from a combination of genes types.
Type I, or “pure” susceptibility genes (herein referred to as S) are defined as genes that increase the risk of schizophrenia but do not preferentially impact any of its associated clinical features. Therefore, relatives inheriting risk alleles in these genes, who as a result became ill, would be uncorrelated for any clinical features such as age of onset, course, or symptomatic profile. Furthermore, in cases due solely to such S genes, clinical features in a proband would be unrelated to the risk of illness in relatives. While clinical features could be correlated in relatives for environmental reasons, such correlations would not differ in MZ vs DZ twin pairs.
Type II, or mixed susceptibility/modifier gene (herein referred to as MS). This class of genes would increase the susceptibility to schizophrenia, but to certain presentations more than others. Examples might include dysbindin and COMT as outlined above. This type, in contrast to S genes, would lead to a greater risk of illness in the relatives of subjects with clinical features associated with the gene in that having that clinical feature would be a result of having a particularly high liability to illness. These features would also be correlated within affected relative pairs, and furthermore, would be associated with greater MZ than DZ twin correlations.
Type III, or “pure” modifier genes (herein referred to as M). These genes would affect the clinical features of the illness, but would not increase susceptibility to illness by themselves. Clinical features influenced by these genes would therefore not predict the risk of illness in relatives, a feature shared with S genes. However, M genes, like MS genes and in contrast to S genes, would cause the resemblance of relative pairs for clinical features, as well as greater MZ than DZ correlations for the same features.
We can discern two more models of gene action to account for effects on nonpsychotic relatives. Type IV is defined as MS genes that influence not only the liability to subtypes of schizophrenia in which specific features are prominent but, in addition also influence attenuated versions of the same features in nonpsychotic relatives. We henceforth call these MS
B (for broad) genes. These would lead to the same study outcomes as MS genes, but additionally would also lead to clinical features in psychotic subjects predicting similar but attenuated features in nonpsychotic relatives, for example, as described by our group (
31). Finally, Type V is denoted as M genes that act broadly, influencing clinical features in psychotic, as well as related traits in nonpsychotic individuals, herein referred to as M
B genes. The effects of these genes on study out-come would be the same as M genes. Additionally, like MS
B genes, M
B genes again would lead to a correlation of certain clinical features in affected subjects with attenuated versions in their relatives. Predictions of outcome in the different types of family, twin, and linkage designs from the five gene types are presented in Table 1.
We would like to note that the actual biological functions of putative susceptibility genes, and therefore, the consequences for brain structure and function of risk alleles in them are only vaguely understood. There are few published studies of the neurobiology of such genes in schizophrenia. These have reported altered expression of both dysbindin (
46) and neuregulin-1 isoforms (
47) in the prefrontal cortex of schizophrenic patients, as well as reduction of dysbindin in presynaptic hippocampal sites (
48). It may therefore seem somewhat premature to describe hypothetical models of gene action without being able to invoke actual neurobiological consequences. However, it could be heuristically valuable to be mindful of the possibility of heterogeneous gene action as we search for new and more powerful linkage and association methods to identify new genes and to validate existing candidates.
Implications for linkage studies
Since affection in complex diseases such as diabetes, hypertension, and hypercholesterolemia is defined quantitatively, it is intuitively appealing that quantitative linkage approaches might have more power to detect susceptibility genes. Simulation studies suggest that continuous traits provide substantially more power to detect genetic effects than do binary or ordinal traits in twin studies (
49). If personality traits in unaffected relatives represent the expression of genes relevant to the etiology of schizophrenia, including these traits in joint analyses could increase the power of linkage studies by including more genetic “information” in analysis. Linkage studies of hypertension (
50) and non-insulin-dependent diabetes mellitus (
51) are encouraging in their successful use of quantitative phenotypes.
In schizophrenia, linkage analysis using quantitative traits can take two fundamentally different approaches, as exemplified by two published studies. Brzustowicz
et al. reported linkage to chromosome 6p using as a quantitative phenotype the positive subscale of the Positive and Negative Syndrome Scale in which
both affected and unaffected relatives were rated (
52). However, there was no evidence for linkage using traditional categorical phenotypes. Wilcox
et al. published a genome-wide scan in which they used factor-derived scores on the positive, negative, and disorganization factors of the Scale for the Assessment of Positive Symptoms and Scale for the Assessment of Negative Symptoms as quantitative phenotypes in which only psychotic subjects were rated (
53). They reported suggestive linkage to chromosomes 6p, 6q, 9p, 12q, and 20q.
This difference is emblematic of the current lack of consensus in the field concerning the appropriate use of clinical features in general, and quantitative traits in particular, in linkage studies of schizophrenia. It is such a fundamental difference that it is difficult to imagine that these two approaches would detect the same types of genes. As gene-finding studies expand beyond the use of traditional operational definitions of affection, it will be essential to have a framework for designing studies and interpreting their results.
The five gene types outlined above would be expected to result in different outcomes in linkage studies. It is obvious that S genes could be detected using categorical phenotypes. However, as these genes have no effect on clinical features, using any of these features as quantitative traits in affecteds only would not yield a linkage signal. However, jointly analyzing affected and unaffected relatives using a trait common to both, as in the study by Brzustowicz et al., could lead to evidence of linkage. Psychotic subjects would have higher trait scores than nonpsychotic ones. Therefore, among affected relative pairs, there would be a correlation between phenotypic similarity and sharing alleles identical by descent at risk loci. This therefore in essence recaptures the affected relative pair linkage method, only replacing a categorical definition of affection with a continuous measure that is highly correlated with it. It is only because of this correlation that linkage to the continuous measure can be detected, and not because the S gene has an influence on this measure that is independent from its influence on illness liability.
On the other hand, both MS and M genes could be detected using a quantitative trait including only affected relative pairs. However, MS genes would additionally be expected to produce a linkage signal at the same locus using a categorical phenotype. M genes on the other hand, having no effect on illness susceptibility, would not be detectable using traditional linkage with a categorical phenotype, but only with a quantitative trait. MSB and MB genes could also be detected using a quantitative trait (such as delusions) in affected subjects only. However, increased power to detect both types of genes would result if unaffected individuals were jointly analyzed using a corresponding “milder” trait (such as magical thinking).
Over and above illuminating the genetic architecture of psychotic illness, understanding mechanisms of gene action will be necessary in order to develop effective treatments once genes are identified. If schizophrenia is indeed caused by aberrant fetal neurodevelopment (
54,
55), reversing the resulting pathology after illness onset may be difficult. However, schizophrenic symptoms are treatable, and increasingly so since the advent of atypical antipsychotics (
56). Therefore, designing treatments based on, for example, the products of modifier genes influencing symptoms could potentially be a more successful alternative.
Future directions
Despite years of pessimism, the first generation of linkage and association studies in schizophrenia has succeeded in identifying replicated susceptibility genes. Another generation will be necessary for a more complete genetic dissection of the illness, taking into account some of the issues raised here. It would be beneficial to the field if existing samples were used to test for linkage and association to clinical features of illness as well as clinically homogeneous subforms of illness. In addition, there are no published genome scans of schizophrenia-related personality traits in unaffected subjects in existing samples. Besides, while a few published studies of the effect of risk alleles in COMT on cognition in normal subjects lend credence to it as a susceptibility gene, there are currently no such studies published of risk alleles in dysbindin, neuregulin-1, RGS4, and other putative susceptibility genes. However, this approach may help validate these alleles as susceptibility factors, and could provide leads to understanding their mechanisms of risk. It may also offer clues to facilitating the identification of as yet undiscovered susceptibility genes.
Finally, continued advancements in methods of correlating genome scan results are needed. One such method, Genome-Scan Meta-Analysis (
57), has been used successfully to examine results across samples in both bipolar disorder (
58) and schizophrenia (
59). However, we currently do not have satisfactory methods for correlating genome scans of different phenotypes in nonoverlapping groups of subjects
within samples. This could be a very useful method of establishing genetic correlations (or discontinuities) between different phenotypes— either distinct subforms of illness or illness and subclinical phenotypes. Bivariate analysis has been performed using twin data to establish genetic overlap between major depression and neuroticism (
45,
60), bipolar disorder (
61), generalized anxiety disorder (
62), and alcohol (
63) and marijuana (
64) use disorders. As twin study methods are based on differences in phenotypic correlations between monozygotic compared to dizygotic twins, they are sensitive only to the aggregate effects of many genes (
65). Testing for genetic correlations using genome scan data could additionally provide specific genomic regions where there are likely to be genes commonly influencing more than one phenotype. This could identify, for example, regions that could be followed up with association studies using samples composed of different but jointly linked phenotypes, thereby possibly increasing statistical power.