The ultimate promise of pharmacogenetics is the possibility that knowledge of a patient’s DNA sequence might be used to enhance drug therapy to maximize efficacy, to target drugs only to those patients that are likely to respond and to avoid adverse drug reactions. The subsequent discussion will briefly review the process by which the disciplines of pharmacogenetics and pharmacogenomics have developed, and then turn to challenges associated with the “translation” of these disciplines from the research laboratory to the bedside, with the eventual goal of developing truly individualized drug therapy.
Pharmacogenetics to pharmacogenomics
The concept that inheritance can have an important role in individual variation in drug response originally grew out of clinical observations of large differences among patients in their response to “large” doses of a drug. Attempts to understand that variation led to twin studies that demonstrated that plasma concentrations or other pharmacokinetic parameters are highly heritable for some drugs (
8,
9), as well as the simultaneous discovery of large variations in drug levels or metabolism that were inherited as Mendelian traits. Many of those early examples, and many of the most striking examples even today, involved pharmacokinetic factors—that is, factors that influence drug concentration. When a patient takes a drug, that drug must be absorbed, distributed to its site of action, interact with its target and, finally, undergo metabolism and excretion (
10).
The majority of “classic” pharmacogenetic traits have involved drug metabolism. For example, one such trait, which was recognized half a century ago, is inherited variation in
N-acetylation, now known to be due to polymorphisms in the
N-acetyltransferase-2 (
NAT2) gene (
11). Genetic variation in
NAT2 is responsible for phenotypic variation in the pharmacokinetics—and, therefore, the effects— of drugs as disparate as the antihypertensive hydralazine, the antiarrhythmic drug procainamide and the antituberculosis agent isoniazid (
12–
14). The effect of
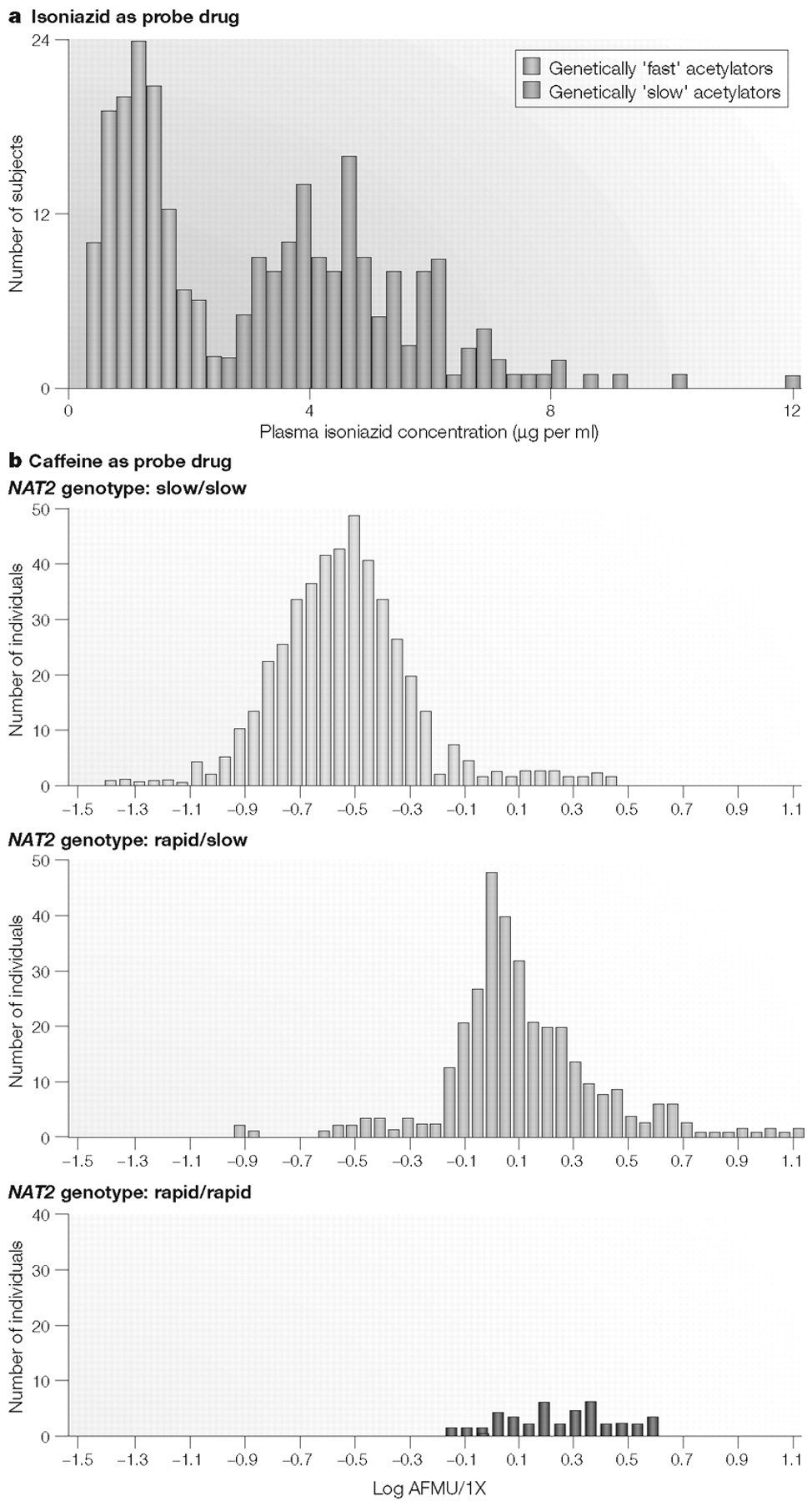
NAT2 pharmacogenetics on plasma levels of isoniazid is shown in Fig. 1A. The bimodal frequency distribution shown in Fig. 1A illustrates the effects of genetically “rapid” acetylation (low plasma drug levels) and genetically “slow” acetylation (high plasma drug levels) (
15). Many early examples of pharmacogenetic variation in drug metabolism involved the measurement of this type of phenotype: plasma drug concentrations, urinary drug excretion, peak plasma levels, drug half-life and so on.
In effect, isoniazid was used as a “probe drug” for
NAT2 polymorphisms to generate the data depicted in Fig. 1A; the plasma concentration of isoniazid provided an indirect reflection of the effects of sequence variation in the gene encoding NAT2, which catalyses isoniazid metabolism (
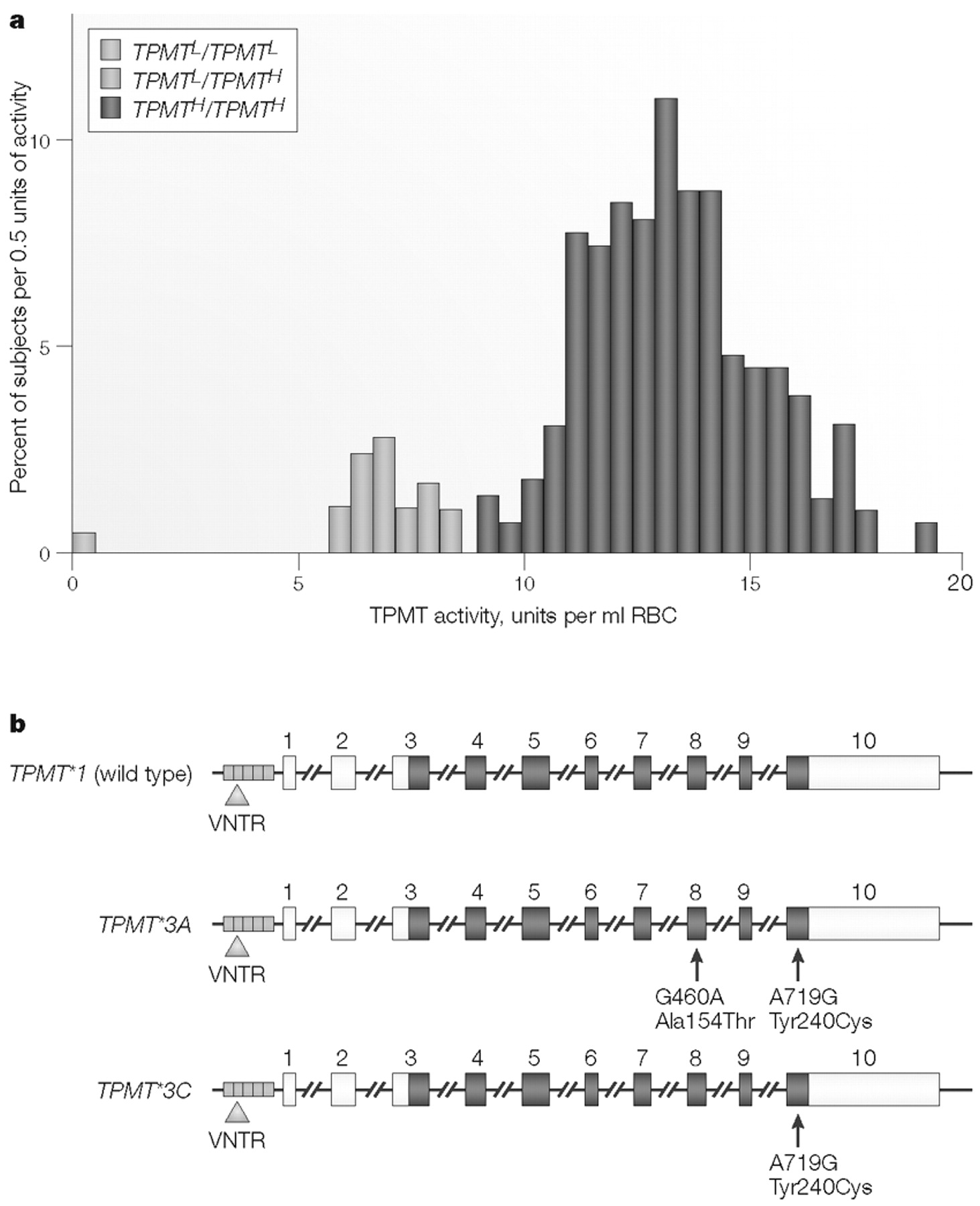
11). However, as shown in Fig. 1B, in which the NAT2 phenotype has been determined with caffeine as the probe drug, genotype and phenotype do not correlate perfectly—a lesson to be remembered whenever DNA-based testing is used in a clinical setting. As described subsequently, “probe drug assays” such as those shown in Fig. 1A,B have been a commonly used pharmacogenetic research tool, beginning at a time before any of the cDNAs or genes encoding proteins responsible for the phenotype being measured had been cloned or characterized. A slightly different approach, involving the assay of a different phenotype, is represented by the original studies of another “classic” example of pharmacogenetics, the thiopurine
S-methyltransferase (
TPMT) genetic polymorphism (Fig. 2A) (
16,
17). In the case of TPMT, the phenotype studied was the level of this drug-metabolizing enzyme activity as measured in an easily accessible cell type, the red blood cell (RBC) (
16,
17). Because the
TPMT genetic polymorphism is of such striking clinical significance, it is described in detail in Box 1 as an example of this type of pharmacogenetic trait.
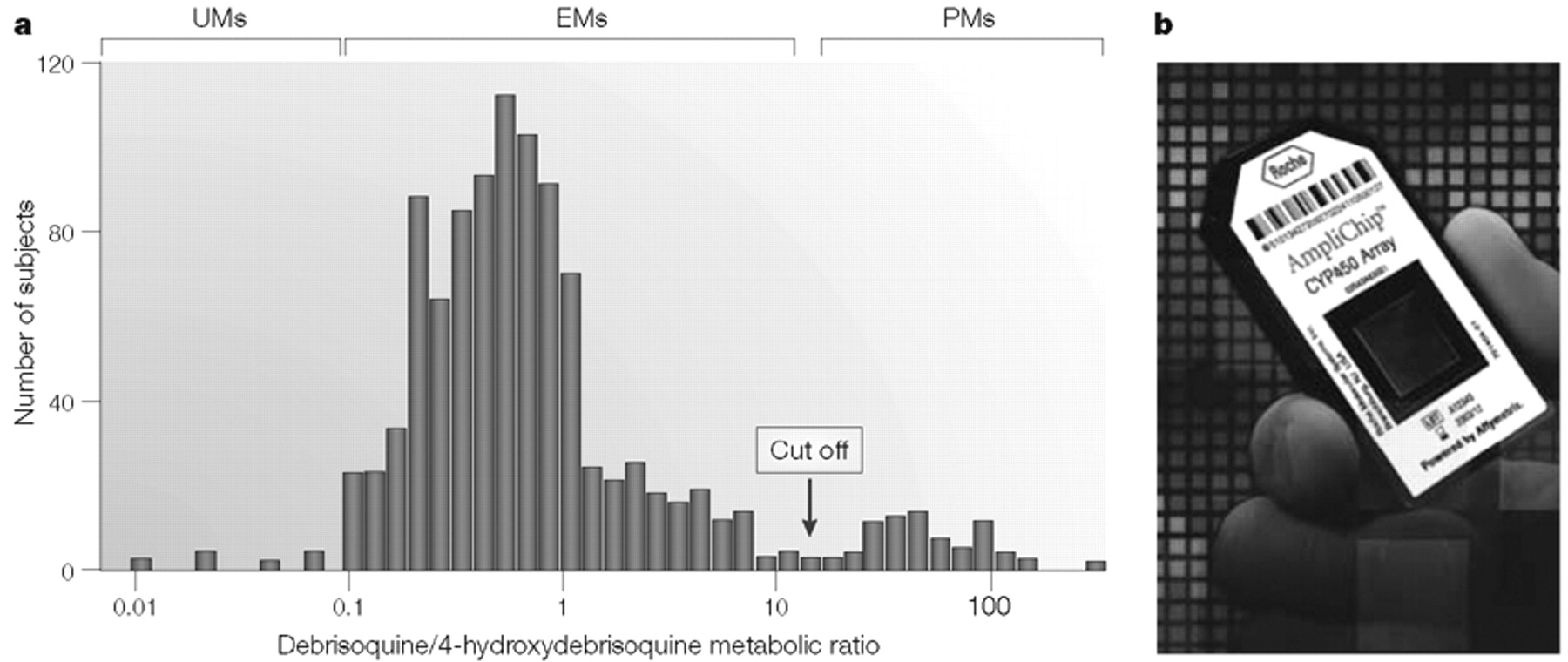
The use of a “probe drug” assay as illustrated for
NAT2 in Fig. 1A,B, or by the administration of drugs such as
debrisoquine to determine cytochrome P450 2D6 (CYP2D6) phenotype (Fig. 3A) (
18–
22), was a mainstay in pharmacogenetic research in the late twentieth century. The frequency distribution depicted in Fig. 3A shows that this Northern European population sample included a group of “poor metabolizers” (PMs) for debrisoquine, a large group of “extensive metabolizers” (EMs) and a small number of “ultra-rapid metabolizers” (UMs), some of whom have been shown to have multiple copies of the
CYP2D6 gene (
23). These UM subjects can display an inadequate therapeutic response to treatment with “standard” doses of drugs metabolized by CYP2D6. Although the occurrence of this phenomenon is relatively infrequent among Northern Europeans, such as the subjects used to obtain the data shown in Fig. 3A, in East African populations the frequency of
alleles with
CYP2D6 gene duplications can be as high as 29% (
24). The frequency distribution histograms of
CYP2D6 and
NAT2 phenotypes that are shown in Figs. 1, 3 have become “icons’ of pharmacogenetics, reproduced in countless textbooks and articles, including the present review (
15,
20)! However, the same probe drug assays used to generate these frequency distributions have also been a barrier to the rapid translation of pharmacogenetics into the clinic. Physicians resisted the requirement that a probe drug be given to patients, and a sample of urine or plasma be obtained, before administration of the desired therapeutic agent. It was at this point that advances in genomic science offered a potential solution to this practical problem and, as a result, an opportunity to help move pharmacogenetics to the bedside.
The application of DNA-based assays in pharmacogenetics promises to make DNA sequence information available to the physician on a timescale such that it can be used practically to help select the best drug and/or dose for each patient. That possibility is indicated symbolically in Fig. 3, in which Fig. 3A shows CYP2D6 phenotype data after the administration of the probe drug debrisoquine and Fig. 3B shows a photograph of a cytochrome P450 microarray that can be used to genotype selected CYP genes, including CYP2D6. The data shown in Fig. 3A, and those generated by the device shown in Fig. 3B, both provide insight into variation in drug response, but the DNA-based technology is potentially faster and requires only a single blood sample without the need for prior administration of a probe drug.
However, it must be acknowledged that our present lack of comprehensive knowledge of genotype-phenotype correlations represents a limitation of the application of genotyping for pharmacogenomic decision making. The phenotype is what the physician wants to know and, unfortunately, present DNA-based tests can fail to reflect the full range of phenotypic variation. As a result, a major challenge for companies designing DNA-based tests is to develop dependable, economical, high-throughput genotyping platforms, and a major challenge for pharmacogenomic science is to determine comprehensive, clinically useful genotype-phenotype correlations.
The
NAT2,
TPMT and
CYP2D6 genetic polymorphisms behave as monogenic Mendelian traits, as do many other “classic” examples from pharmacogenetics. These relatively simple, but striking, examples helped to provide the foundation for our present understanding that inheritance can play an important role in individual variation in drug response by influencing efficacy, toxicity or both. Many additional examples have continued to accumulate in recent years. However, in its 2003 draft “Guidance for Industry Pharmacogenomic Data Submissions” (
25), the US FDA singled out as examples of “valid biomarkers” for pharmacogenomics only the
CYP2D6 and
TPMT polymorphisms— both of which were originally described approximately a quarter of a century ago (
16,
18,
26). The FDA definition of a valid biomarker is one for which an established and validated assay exists and—most important—for which an established body of evidence exists that supports its pharmacological and/or clinical significance (
25). Among the challenges facing pharmacogenomics is how to move beyond
CYP2D6,
TPMT and other classical genetic polymorphisms to broaden the discipline and to move this knowledge from the research laboratory to the patient care environment.
The
NAT2,
TPMT and
CYP2D6 polymorphisms—as well as a series of similar monogenic pharmacogenetic traits—represent easily understood examples that helped to establish that inheritance is an important factor accounting for individual differences in drug response. They served to stimulate the development of the discipline, but even the
TPMT and
CYP2D6 polymorphisms fail to explain all variation in response to drugs metabolized by these enzymes—nor would anyone who has ever written a prescription expect that a single factor would be able to explain all variation in such a complex phenotype. Therefore, to state the obvious, no pharmacogenetic trait, and no test for that trait, should be expected to explain all the observed variation in drug response. For example, there are many reasons why patients with leukaemia who are treated with thiopurine drugs as well as other cytotoxic agents might develop myelosuppression, and a genetically low level of TPMT is only one of those reasons (Box 1). However, if a patient is homozygous for
TPMT*3A, the evidence is now overwhelming that their physician should anticipate significant and perhaps life-threatening myelosuppression in response to treatment with standard doses of thiopurine drugs (
17,
27–
29).
The pharmacogenetic examples cited so far have all involved drug-metabolizing enzymes that influence drug pharmacokinetics, but there are increasing numbers of examples of striking pharmacogenomic variation that influence pharmacodynamics as a result of inherited variation in drug targets. Most pharmaceutical companies now attempt to avoid developing drugs that are metabolized primarily by polymorphic enzymes such as CYP2D6. However, even though it might be possible to minimize the impact of genetic variation on drug metabolism and transport—that is, pharmacokinetic variation—it will be much more difficult to avoid inherited variation in drug targets.
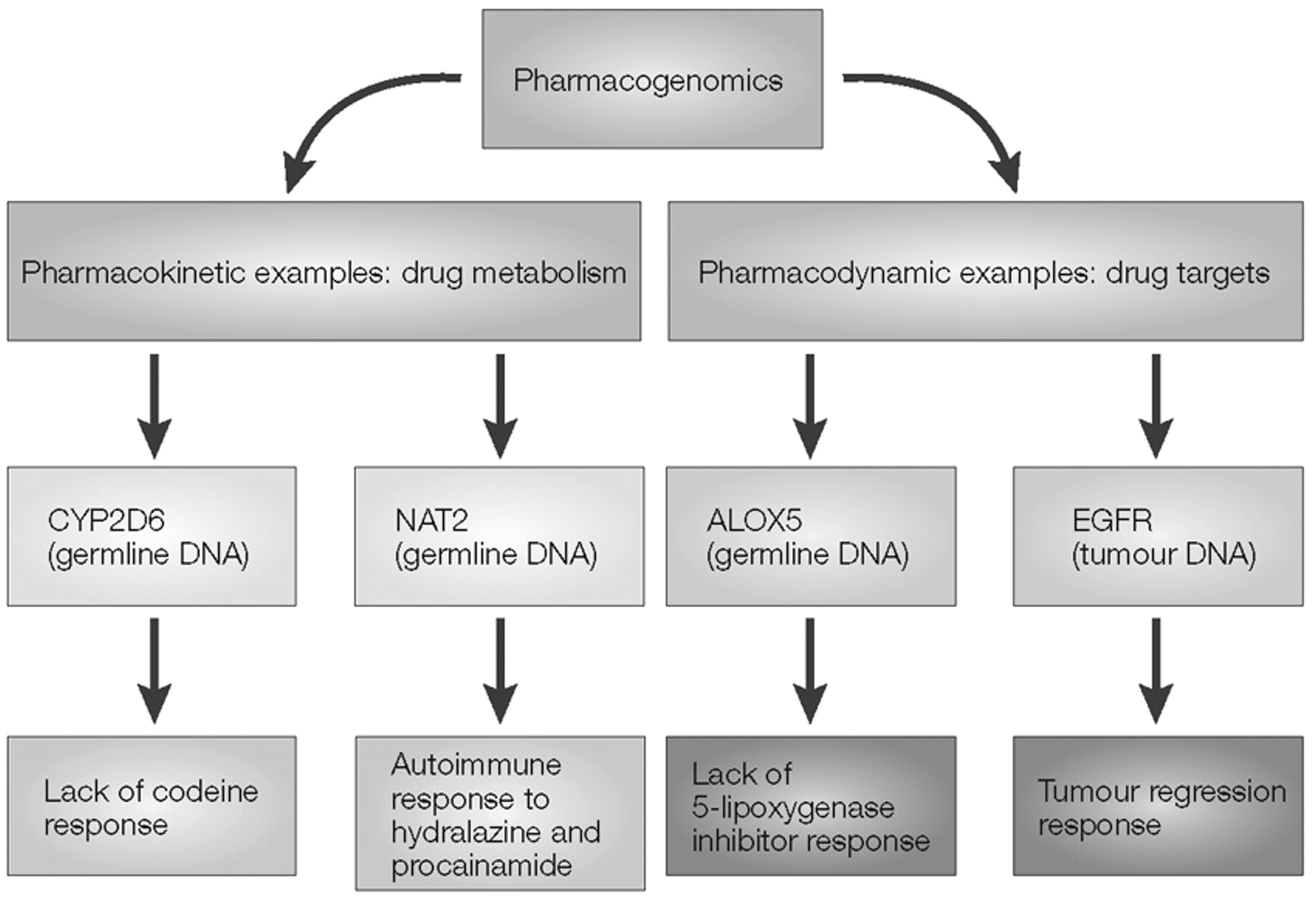
The contrast between pharmacokinetic and pharmacodynamic pharmacogenomic effects is outlined schematically in Box 2, which illustrates two examples of polymorphic enzymes that result in pharmacokinetic variation, CYP2D6 and NAT2. Those two examples are contrasted with two “pharmacodynamic” examples, the
ALOX5 gene that encodes 5-lipoxygenase and the epidermal growth factor receptor (
EGFR) gene. As described in more detail in Box 2, subjects with a variant
variable number of tandem repeats (VNTRs) in the
ALOX5 promoter have decreased transcription of the gene (
30) and, as a result, respond less well to treatment with the 5-lipoxygenase inhibitors that are used to treat asthma (
31). On the other hand, mutations in the
EGFR gene in tumour DNA in non-small-cell lung carcinomas, all occurring within the ATP-binding pocket of the tyrosine kinase domain of this receptor, are associated with enhanced tumour response to the EGFR tyrosine kinase inhibitor gefitinib (Iressa; AstraZeneca) (
32,
33). In these studies, the frequency of these EGFR mutations varied from as low as 2% in patients in the United States to 26% in patients in Japan (
32,
33).
These examples of genetic variation in drug targets might be representative of a large part of the “future” of pharmacogenomics—a future in which, before therapy, patients will be stratified on the basis of their ability to respond or not respond to a therapeutic agent. However, this future scenario will have significant economic implications for the pharmaceutical industry, as discussed subsequently.
The development of pharmacogenetics occurred in parallel with rapid changes in genomic science, most significantly the conception, implementation and completion of the Human Genome Project (
1,
2). At the end of the twentieth century, the convergence of these two areas of biomedical research resulted in the evolution of pharmacogenetics into pharmacogenomics (
34,
35). Although it might seem that there are nearly as many definitions of “pharmacogenomics” as there are investigators engaged in the discipline, the terms “pharmacogenetics” and “pharmacogenomics” are often used interchangeably. From the perspective of the authors of this review, pharmacogenomics emerged from the convergence of the step-wise advances that occurred in pharmacogenetics during the twentieth century with the striking changes that occurred in genomic science at the end of that century, such as the completion of the Human Genome Project, and the development of expression profiling, as well as high-throughput DNA sequencing and genotyping (see Refs.
34,
35 for recent reviews).
Whatever definition of pharmacogenomics one might choose to use, the latter portion of the twentieth century witnessed the emergence of the concept that inheritance is a major factor responsible for variation in drug response. Once that principle had been established, the question immediately arose of the best way by which to translate this information to the bedside. Furthermore, as the twentieth century ended, that question was being asked within the context of rising enthusiasm for all things “genomic”—an enthusiasm that might have led to unrealistic expectations with regard to our ability to “individualize” drug therapy on the basis of genomics. Those unrealistic expectations might have occurred in part because of the understandable enthusiasm of investigators in this area of research; in part because of naivete with regard to the difficulty of the clinical validation and acceptance by practicing physicians of laboratory-based observations; and, in part, because of a need to raise venture capital by start-up biotechnology firms that were attempting to commercialize pharmacogenomics.
Regardless of the reasons, the fact is that although pharmacogenomic testing had been predicted to be one of the first broad applications of genomics to clinical medicine (
36)—and this might ultimately prove to be correct—such applications so far have been limited to a few tests that are used mainly within academic referral centres. The relatively slow pace of the incorporation of pharmacogenomics into clinical practice has, in turn, resulted in impatience and even disillusionment with regard to the clinical potential of this area of biomedical science (
37). Therefore, the questions of why the pace has been so “measured,” and what might be done to accelerate the translation of this body of knowledge to the bedside, need to be addressed. The subsequent discussion will attempt to briefly outline some of the challenges that exist as we attempt to move pharmacogenetics and pharmacogenomics into the clinic, as well as issues that will have to be addressed if that process is to be accelerated.