Seeking food and companionship and avoiding physical and psychological discomfort are examples of motivated adaptive behaviors. Motivated behavior classically implies both an activation of the organism by environmental or interoceptive stimuli and a directed behavioral output (
10). Thus, the neurobiological search for the antecedents of motivated behavior involves defining the neural substrates that 1) attach sufficient importance (salience) to an integrated stimulus that behavior is “activated” and 2) “direct” this state of activation toward a specific behavioral response. While we have made substantial progress toward identifying the neural circuits and cellular foundations responsible for activating behavior, we have been only marginally effective at understanding the substrates that cause one behavior to be favored over another behavior (direction of behavior).
ACTIVATION OF BEHAVIOR
Neurobiology has focused on three brain regions in the activation of behavior: the amygdala, prefrontal cortex, and nucleus accumbens. The amygdala emerged from studies showing involvement in fear-motivated behaviors (
11), while the nucleus accumbens was identified from a connection with reward-motivated behaviors (
12). The prefrontal cortex is less involved in establishing whether a stimulus is positive or negative (valence); rather, it regulates the overall motivational salience and determines the intensity of behavioral responding (
13,
14). More recent studies have blurred the linkage between positive and negative emotional valence in the amygdala and nucleus accumbens, and they have revealed a neuronal circuit consisting of glutamatergic interconnections among the amygdala, nucleus accumbens, and prefrontal cortex and dopaminergic afferents to all three regions (
15,
16).
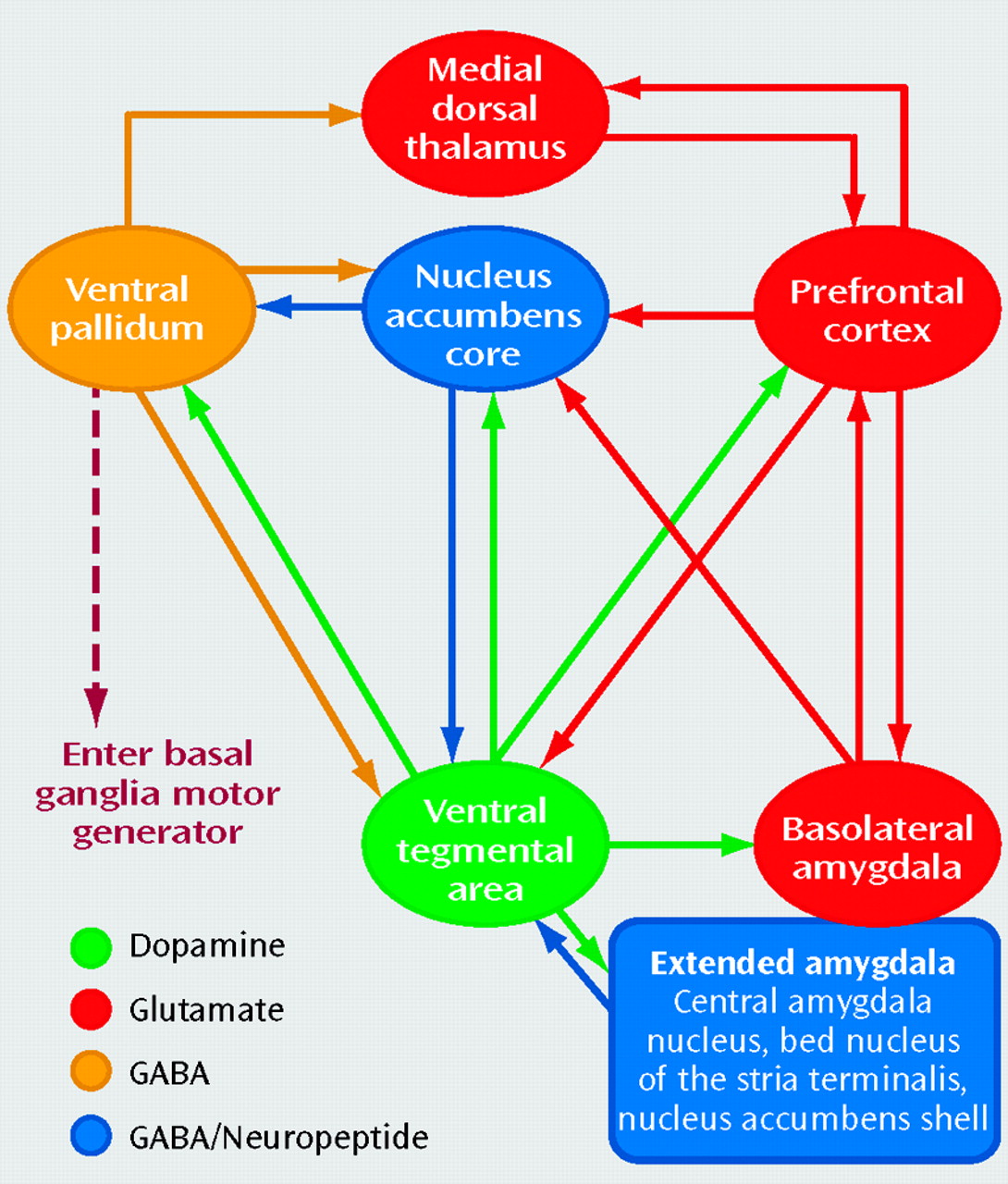
Figure 1 illustrates this circuit and includes three additional components. The accumbens has dense projections carrying γ-aminobutyric acid (GABA) and neuropeptides to the ventral pallidum that are critical for the expression of motivated behaviors (
17). Another GABA/neuropeptide component of the circuit is the extended amygdala, which is a cluster of interconnected nuclei, including the central amygdala nucleus, bed nucleus of the stria terminalis, and shell of the nucleus accumbens, that is in part a conduit for environmental and interoceptive stressors (
18). It is important to note that while the shell of the accumbens possesses some functional and anatomical characteristics of the extended amygdala (especially in terms of the neurocircuitry of addiction), it is also anatomically distinct from the other nuclei in terms of some aspects of connectivity and histochemistry (
19). Finally, there is a series subcircuit consisting of GABA-ergic projections from the ventral pallidum to the mediodorsal thalamus and a reciprocal glutamatergic projection between the thalamus and prefrontal cortex that mediates reintegration of information exiting the circuit back into the prefrontal cortex (
20).
Dopamine and the ventral tegmental area. Projections from the ventral tegmental area release dopamine throughout the circuit in response to a motivationally relevant event (
21,
22). The release of dopamine signals the circuit to initiate adaptive behavioral responses to the motivational event, and in doing so it facilitates cellular changes that establish learned associations with the event (
5). In this way the organism can more effectively emit an adaptive behavioral response should the event reoccur. However, in contrast to repeated drug administration, as a motivational event becomes familiar by repeated exposure, dopamine release is no longer induced by that particular event (
23). In this case, although the behavioral response remains goal directed, it is well learned and further dopamine-induced neuroplastic changes are not necessary. However, it is important to note that conditioned stimuli predicting the event continue to trigger release of dopamine (
23–
25). Therefore, in most natural situations where learned associations accompany a repeatedly encountered motivational event, dopamine will likely be released as part of the overall experience. In sum, dopamine can be seen as serving two functions in the circuit: 1) to alert the organism to the appearance of novel salient stimuli, and thereby promote neuroplasticity (learning), and 2) to alert the organism to the pending appearance of a familiar motivationally relevant event, on the basis of learned associations made with environmental stimuli predicting the event (
23,
26).
Amygdala. The amygdala is especially critical in establishing learned associations between motivationally relevant events and otherwise neutral stimuli that become predictors of the event (
27). Interactions between the basolateral and central amygdala nucleus involve autonomic and endocrine associations by means of projections from the central nucleus to the brainstem, the hypothalamus, and dopamine neurons in the ventral tegmental area (
28,
29). In contrast, the glutamatergic projections from the basolateral amygdala to the prefrontal cortex and accumbens are required for learned associations to influence more complex behavioral responses (
16,
20). The functional integration between the amygdala and prefrontal cortex has been demonstrated in many neuroimaging studies in which healthy subjects were exposed to stimuli associated with motivationally relevant events, ranging from food and sex to social cooperation (
30–
32).
Prefrontal cortex. The anterior cingulate and ventral orbital cortices in the prefrontal cortex are recruited by motivationally relevant events, as well as stimuli that predict such events, and contribute to whether a behavioral response will be emitted and the relative intensity of that response (
14,
33). Consistent with involvement of dopaminergic afferents, the activation of the prefrontal cortex by rewarding stimuli is strongly influenced by the predictability of the reward (
34,
35).
Nucleus accumbens. The accumbens contains two functionally distinct subcompartments, termed the shell and core (
36). The shell is strongly interconnected with the hypothalamus and ventral tegmental area and is correspondingly important in regulating ingestive behaviors (
21,
36). Reciprocal dopamine innervation from the ventral tegmental area to the accumbens shell is important in modulating motivational salience and contributes to establishing learned associations between motivational events and concurrent environmental perceptions (
37,
38). In contrast, the core compartment is anatomically associated with the anterior cingulate and orbitofrontal cortex and appears to be a primary site mediating the expression of learned behaviors in response to stimuli predicting motivationally relevant events (
36,
39). Moreover, the obligatory involvement of the accumbens core in expressing adaptive behavior depends not on dopaminergic afferents but, rather, on glutamatergic afferents from the prefrontal cortex (
40). Although not an obligatory event, dopamine is released into the core in response to stimuli predicting a rewarding event and likely modulates the expression of adaptive behaviors (
41,
42).
DIRECTION OF BEHAVIOR
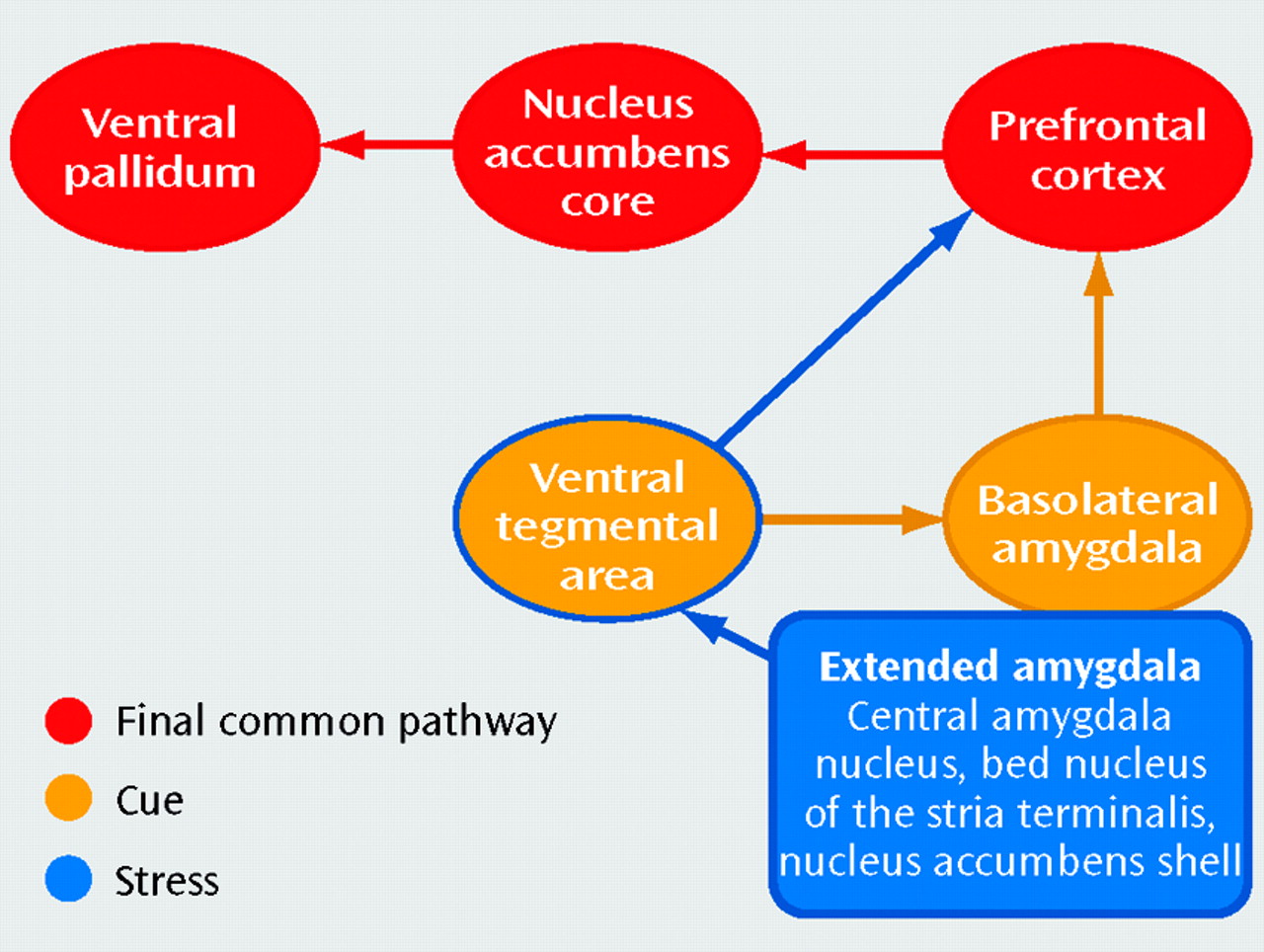
While our understanding of brain mechanisms responsible for activating goal-directed behavior is considerable, the mechanisms by which the circuit in
Figure 1 determines or “chooses” the direction of behavior are less clear. Choice is initiated in part by means of the prefrontal cortex, as some studies have shown that activation of the prefrontal cortex precedes behavior (
33,
43,
44). Glutamatergic efferents from the prefrontal cortex stimulate behavioral output by accessing accumbens-thalamocortical circuitry (
45,
46). It has long been proposed that distinct neuronal ensembles within the accumbens encode the relationship between discrete stimuli and behavioral responses (
47). However, only recently was it demonstrated that different subsets of neurons in the accumbens respond differentially to cues associated with distinct motivationally relevant stimuli, such as water versus cocaine (
48). How these ensembles are formed and organized is unclear. However, the intensity and quality of behavioral output are strongly influenced by both dopaminergic and glutamatergic input to the accumbens, and activity at these synapses produces morphological changes in the dendrites of accumbens spiny cells (
49). Changes in dendritic spine density occur in cellular and in vivo models of learning, and they correlate roughly with excitatory synaptic contacts (
50). In addition to morphological changes, in vitro models of neuroplasticity reveal intracellular changes that can augment or diminish excitatory transmission (
51,
52). Recent studies demonstrate that addiction is associated with neuroplasticity in these cellular mechanisms of synaptic organization, and we will discuss them in detail.