Pathophysiological changes in the brain that are associated with psychiatric disorders or animal models of these conditions include gross differences in the sizes of specific brain regions, alterations in the morphology of subpopulations of neurons, neurochemical changes at the synaptic cleft, alterations in intracellular signalling and changes in the regulation of gene expression. Most psychiatric disorders share important features, including a substantial genetic predisposition (
1)) and a contribution from environmental factors. Another common attribute of psychiatric conditions is long-lasting behavioural abnormalities. In most individuals, these illnesses develop gradually and show a chronic, remitting course, often over a lifetime. Likewise, the reversal of symptoms in response to treatment occurs over weeks or months. Psychiatric medications are virtually unique in their requirement for chronic administration to achieve their full clinical effect. Accordingly, an important challenge in psychiatric research has been to identify the molecular basis of stable changes in behaviour that account for both the symptoms of mental illness and their reversal during treatment.
The regulation of gene expression has been proposed as one molecular mechanism that could mediate stable adaptations and maladaptations in the brain(
2). Changes in mRNA levels have been documented in specific brain regions both in animal models of psychiatric illness and in human brains, and have been related to altered behaviour. However, it has been difficult to identify the molecular mechanisms that underlie such stable changes in gene expression; virtually all reported changes in transcription factors and other nuclear regulatory proteins in animal models revert to normal within hours or days of chronic perturbation. One exception is the induction of ΔFOSB, a FOS family transcription factor, which accumulates in specific regions of the brain in response to many chronic stimuli (drugs of abuse, stress, antipsychotic drugs and so on), and persists for several weeks after the end of the stimulus (
3,
4). But even the induction of ΔFOSB does not persist as long as the behavioural changes. Thus, the search continues for the molecular basis of particularly stable adaptations and maladaptations in the brain.
Recent research has raised the notion that epigenetic mechanisms, which exert lasting control over gene expression without altering the genetic code, could mediate stable changes in brain function. Historically, the field of epigenetics has focused on how cellular traits can be inherited without a change in DNA sequence. Studies of epigenetic mechanisms that underlie heritable transmission have flourished in the fields of developmental and cancer biology, where the continuity of unique patterns of gene expression between parent and daughter cells is crucial. These studies have converged on a set of common enzymatic modifications to chromatin structure that can up- or downregulate gene expression in a manner that is transmissible to daughter cells. These mechanisms also regulate gene expression in neurons, but, as most neurons do not divide, chromatin modifications are instead sustained within individual cells.
This review outlines recent discoveries involving the epigenetic regulation of neurobiological adaptations that are associated with long-lasting behaviours in animal models of psychiatric conditions and in the brains of humans with these disorders. After a brief overview of epigenetic mechanisms, we focus on a small number of conditions, including depression, addiction, schizophrenia and developmental disorders, for which the supporting evidence is best established.
Overview of epigenetic mechanisms
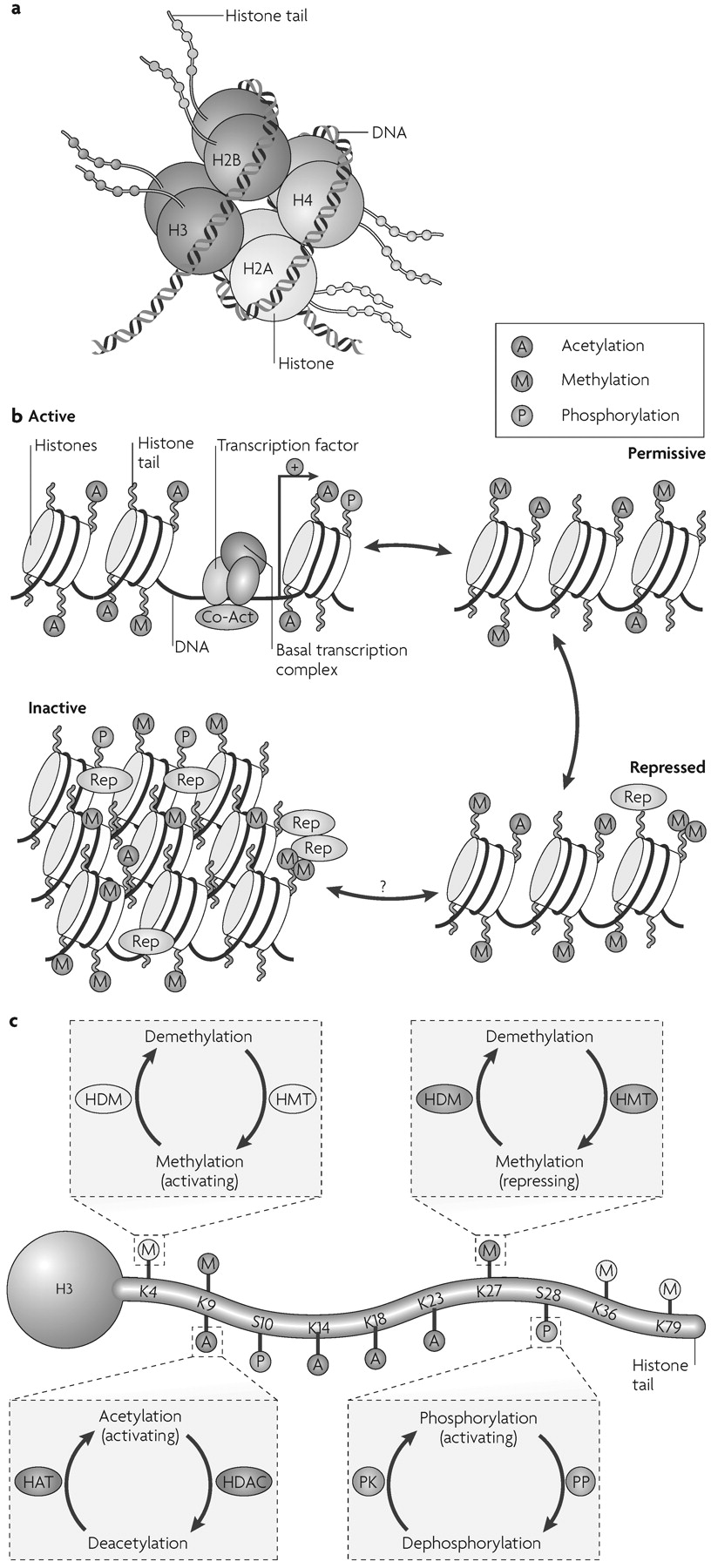
Chromatin is the complex of DNA, histones and nonhistone proteins in the cell nucleus. Remodelling of chromatin is a dynamic process that modulates gene expression. The fundamental unit of chromatin is the nucleosome, which consists of ≈147 base pairs of DNA wrapped around a core histone octamer (≈1.65 turns). Each octamer contains two copies each of the histones H2A, H2B, H3 and H4 (
Fig. 1a). The nucleosomal structure of chromatin allows DNA to be tightly packaged into the nucleus by organized folding (
5). Intricate chromatin remodelling mechanisms ensure that DNA remains accessible to the transcriptional machinery. These epigenetic mechanisms alter gene activity by modulating DNA-protein interactions without changing the genetic code.
In simplified terms, chromatin exists in an inactivated, condensed state, heterochromatin, which does not allow transcription of genes, and in an activated, open state, euchromatin, which allows individual genes to be transcribed (
Fig. 1b). The opening of chromatin is associated with acetylation of nearby histones, although it remains unclear whether acetylation mediates or reflects chromatin decondensation. In reality, chromatin can exist in many states in between these two extremes (
Fig. 1b). Portions of chromatin are highly repressed, owing to DNA and histone methylation and the binding of repressor proteins, and might never be accessible for transcription. Other portions of chromatin are in repressed or permissive states; their basal activity is low owing to histone methylation and perhaps other modifications, but the genes are available for derepression and activation in response to transcription factors and transcriptional co-activators. Chromatin remodelling modulates gene expression with high temporal and spatial resolution by permitting small groups of nucleosomes to become more or less open, which consequently enhances or inhibits access of the transcriptional machinery to specific promoter regions.
Experiments in yeast have yielded detailed information about the molecular mechanisms that control chromatin architecture to alter gene expression. Several general mechanisms have emerged and it is generally believed that their complex interactions determine the appropriate expression of specific genes in eukaryotic cells (
5,
6).
By far the best characterized chromatin remodelling mechanism in the brain is the post-translational, covalent modification of histones at distinct amino acid residues on their amino (N)-terminal tails. Such modifications include acetylation, ubiquitylation or SUMOylation at lysine (K) residues, methylation at lysine or arginine (R) residues, phosphorylation at serine (S) or threonine (T) residues, and ADP-ribosylation at glutamate (E) residues (
Fig. 1c). Hyperacetylation is generally thought to promote decondensation of chromatin and an increase in gene activity, whereas hypoacetylation marks condensation and decreased activity. It has also been proposed that increased gene activity is best associated not with the level of acetylation, but with the dynamic cycling of acetylation and deacetylation. In contrast to acetylation, histone methylation can correlate with either gene activation or repression, depending on the residue undergoing methylation (
7). Phosphorylation of histones is also associated with chromatin inhibition or activation (
6). The roles of histone ubiquitylation, SUMOylation and ADP ribosylation are less well understood (
8,
9). The diversity of histone modifications supports the “histone code hypothesis”, which posits that the sum of modifications at a particular promoter region defines a specific epigenetic state of gene activation or silencing (
10).
The enzymes that mediate these covalent modifications are becoming increasingly understood. Many histone acetyltransferases (HATs), which catalyse acetylation, have been identified. Several transcriptional activators contain intrinsic HAT activity (
10,
11). Histone deacetylases (HDACs) catalyse deacetylation; they also associate with several transcriptional repressors to further repress chromatin activity (
11) (Box 1). The balance between the opposing activities of HATs and HDACs maintains acetylation on core histones and is thought to be an important determinant of transcription. Methylation at lysine or arginine residues is mediated by histone methyltransferases (HMTs). In general, histone lysine methylation is regarded as a more stable modification than other histone modifications, which seem to be more readily reversible (
6,
7), although the recent discovery of histone demethylases (HDMs) indicates that even methylation can be reversed (
12).
Several other general mechanisms of chromatin remodelling have been described, although they remain less well characterized in the nervous system. Nucleosome sliding involves the movement of the histone octamer along DNA and thereby allows the transcriptional machinery to transcribe a gene (
5). This process is facilitated by the SWI/SNF family of chromatin remodelling complexes, which use ATP-derived energy to disrupt nucleosome structure non-covalently (
11). Histone substitution, where canonical histones within a nucleosome are switched with naturally occurring histone variants, is a further example of chromatin remodelling, although its physiological function in the brain is poorly understood (
6).
DNA methylation is another important mechanism of gene repression. It occurs by transfer of a methyl group from S-adenosyl methionine (SAM) to cytosine residues at the dinucleotide sequence CpG, and is catalysed by DNA methyltransferases (DNMTs) (
6,
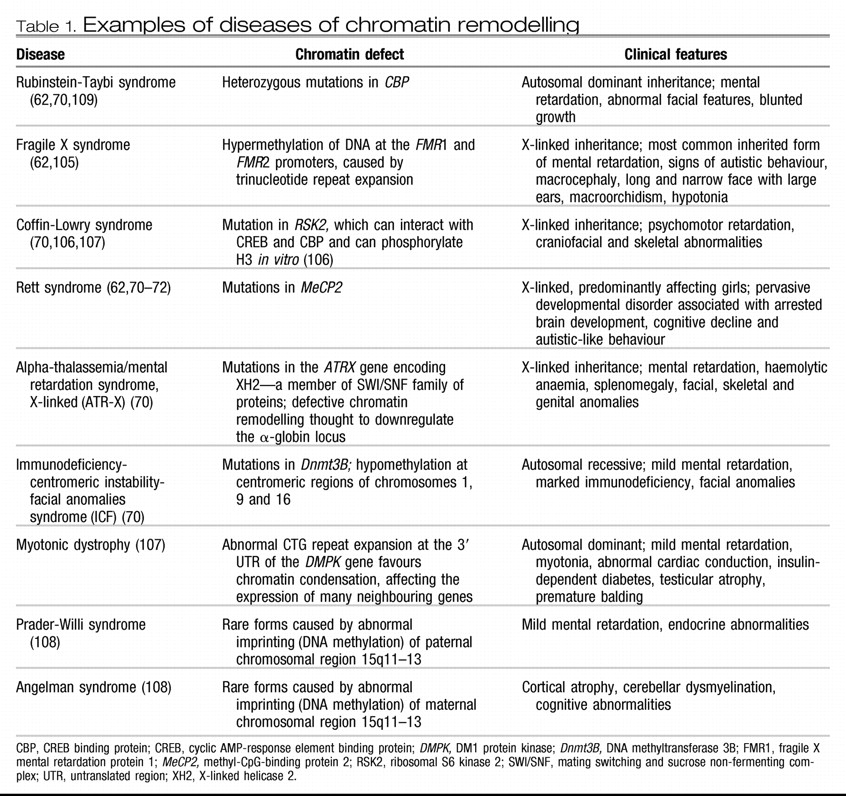
7). Although CpG sequences throughout the genome are usually heavily methylated, those at the promoter regions of genes, specifically at CpG clusters or islands, are methylated to a much lesser extent, and the amount of DNA methylation at a promoter correlates with the extent of gene inactivation. The functional significance of DNA methylation is best established in X chromosome inactivation and genetic imprinting; abnormal imprinting can lead to neurodevelopmental diseases (Box 1;
Table 1). More recently, DNA methylation has been implicated in the regulation of gene activity in the adult brain under normal and pathological conditions (see below).
Patterns of DNA methylation are intricately linked to patterns of histone modification. Methyl binding domain proteins (MBDs), such as methyl-CpG-binding protein 2 (MeCP2), can be recruited to methylated DNA. MBDs are associated with large protein complexes containing HDACs and HMTs, which further repress gene transcription (
7). Thus, DNA methylation and histone methylation and deacetylation are intricately interconnected, each representing epigenetic hallmarks of the silenced promoter.
Signalling pathways in chromatin remodelling
Although chromatin remodelling is best understood for its influence on neural development (Box 1), increasing evidence suggests a role in regulating mature, fully differentiated neurons. During synaptic transmission, neurons respond to neurotransmitters by receptormediated intracellular signal transduction events, which, among other actions, activate or inhibit transcription factors. The regulation of transcriptional activity by transcription factor binding to DNA depends on interactions of the transcription factors with many co-activators or co-repressors and the underlying structure of chromatin. Chromatin remodelling is thus intimately linked to activation or repression of genes by synaptic activity. Such mechanisms regulate the expression of specific sets of neuronal genes that are important for neural activity, survival, morphology and ultimately the integrated regulation of complex behaviour.
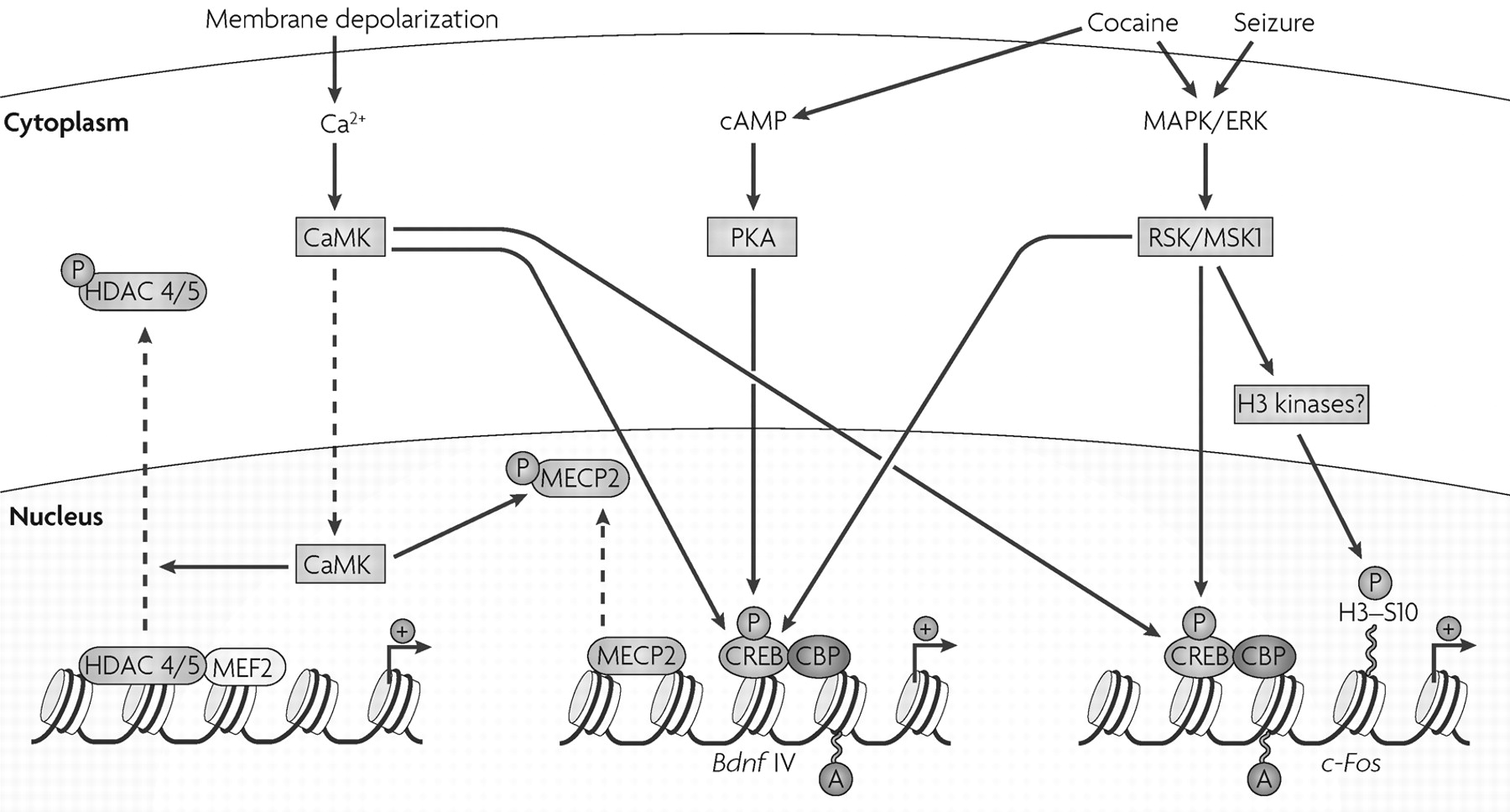
We are beginning to understand how intracellular signalling in the brain regulates chromatin remodelling. The best-established mechanism involves the transcription factor cyclic AMP (cAMP)-response element binding protein (CREB). The activation of several signalling pathways involving cAMP, Ca
2+ and extracellular signal regulated kinase (ERK) leads to the phosphorylation of CREB at Ser133 (
Fig. 2). CREB phosphorylation triggers the recruitment of CREB-binding protein (CBP), a transcriptional co-activator whose intrinsic HAT activity acetylates nearby histones, which loosens the chromatin and allows subsequent transcriptional activation. CBP is important for normal learning and memory, and mutations of CBP cause Rubinstein-Taybi syndrome, a form of mental retardation in humans (see below).
Several external stimuli can induce rapid changes in histone modifications in the brain, but the intracellular mediators of these signals are poorly defined. For example, cocaine and antipsychotic drugs induce acetylation of histone H4 and phosphoacetylation of histone H3 in the striatum (a brain region that is important for the behavioural effects of these drugs; see below) (
13–
15). Among the genes that show the most marked histone changes, which can be identified by use of chromatin immunoprecipitation (ChIP) assays, are immediate-early genes, such as
c-Fos. c-Fos transcription is induced rapidly in the brain by numerous stimuli, including cocaine, antipsychotic drugs and seizures. These stimuli trigger rapid and transient enrichment of H4 acetylation and H3 phosphoacetylation at the
c-Fos promoter in neurons, in association with transcriptional activation of
c-Fos. However, the signalling pathways through which these stimuli modify histones are unknown. One study implicated the ERK pathway, in particular mitogen- and stress-activated kinase 1 (MSK1), in cocaine induction of H3 phosphorylation and
c-Fos activation in the striatum (
13), although it remains unclear whether MSK1 directly phosphorylates H3 or induces its phosphorylation by other kinases. Likewise, the ERK pathway may mediate H3 phosphoacetylation in response to seizures (
16). Several other kinases can phosphorylate H3 in non-neuronal cells, but their actions in the brain are unexplored (
17). Therefore, although H3 phosphoacetylation has been directly associated with
c-Fos induction by acute stimuli, much work is needed to understand the underlying molecular mechanisms involved.
Indeed, one of the main challenges in the field is to elaborate the precise steps through which neural activity and synaptic transmission signal to the nucleus to regulate the enzymes and other proteins that mediate chromatin remodelling. Insight into these pathways has come from studies of non-neural cells. The activation of cellular Ca
2+ pathways in muscle leads to the activation of CaMKs (calcium/calmodulin-activated protein kinases), which phosphorylate class II HDACs. This phosphorylation triggers the shuttling of the enzyme out of the nucleus, and results in increased histone acetylation (
Fig. 2). This pathway has now been demonstrated in hippocampal cells (
18) and cerebellar granule neurons (
19), indicating that chromatin signalling mechanisms in different tissues overlap substantially.
It is unclear how histone acetylation can be regulated only at specific genes, but this is believed to involve multiprotein complexes. For example, class II HDACs can target specific genes for repression through N-terminal regulatory domains that mediate interactions between these HDACs and certain transcription factors, such as myocyte enhancing factor 2 (MEF2) (
Fig. 2). Independent of histone deacetylation, class II HDACs can recruit cyclin-dependent kinase 5 (CDK5) to phosphorylate MEF2 and repress its transcriptional activity (
19,
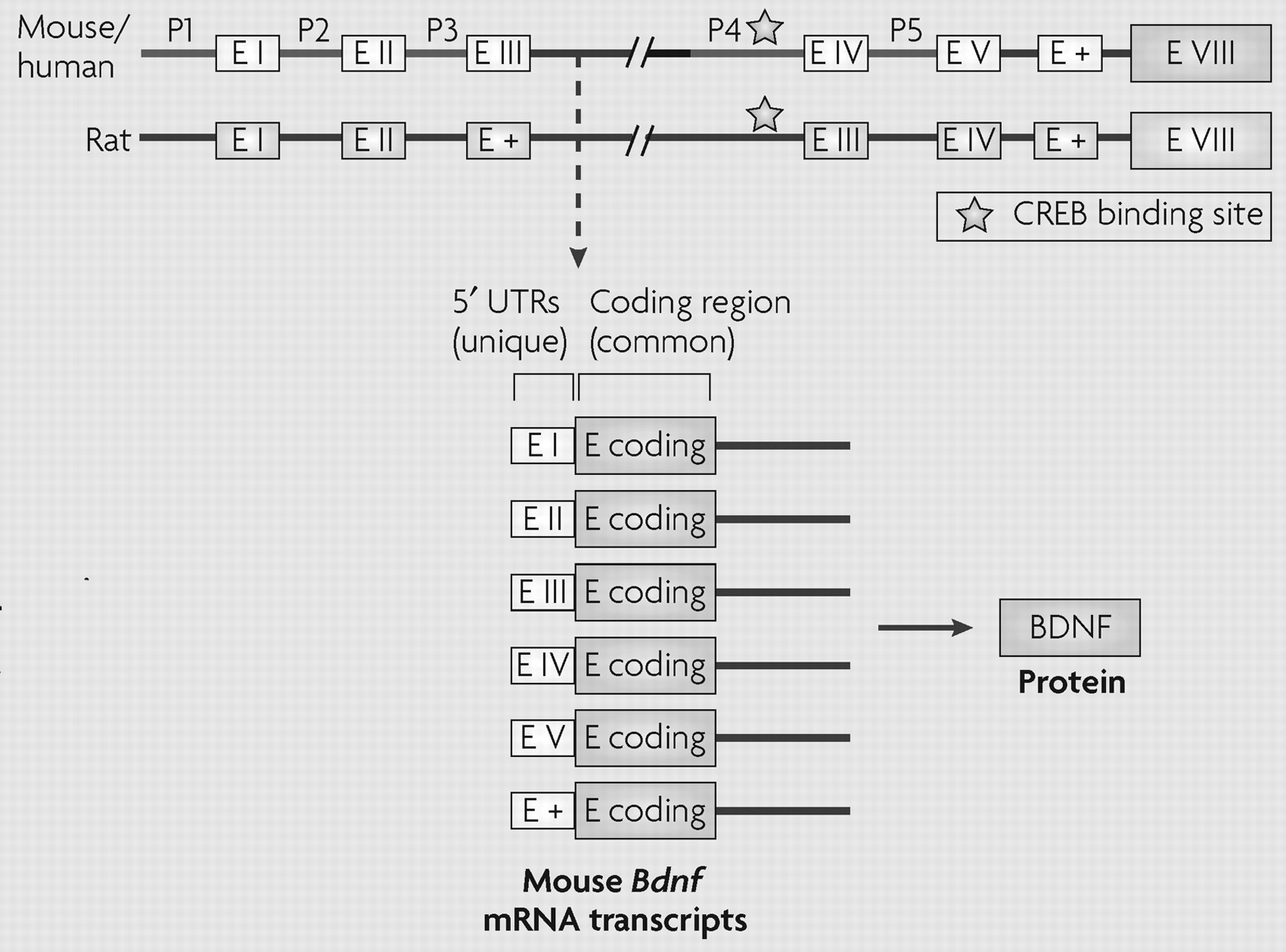
20). However, little is known about these regulatory mechanisms in the brain. Among neural genes, brain-derived neurotrophic factor (
Bdnf) is one of the most studied for its regulation by chromatin remodelling (
Box 2).
The above studies underscore the importance of dynamic chromatin remodelling in the transcriptional response to acute stimuli in neuronal cells. However, much research on psychiatric disorders is focused on neuroadaptations that evolve slowly but can cause lasting changes in brain circuitry. The next sections provide evidence for such stable neuronal regulation at the level of chromatin remodelling as it relates to the pathogenesis and maintenance of complex psychiatric disorders.
Epigenetic mechanisms in depression
Depression is a common, chronic and debilitating disease. Although many patients benefit from antidepressant medication, electroconvulsive seizures (ECS) or psychotherapy, only about half of depressed patients show a complete remission, which underscores the need for more effective agents (
21). The mechanisms that precipitate depression, such as stress, are incompletely understood. One mystery of the disease is its longlasting nature and delayed response to antidepressant treatment. This persistence is thought to be mediated by slowly developing but stable adaptations, which might include epigenetic regulation.
To investigate such adaptations, one study examined changes in histone modifications after chronic ECS in the rat hippocampus (
22), a brain region implicated in the pathophysiology and treatment of depression (
23,
24). Like antidepressant medications, ECS is effective only after repeated administration, indicating that long-term adaptations at the level of gene expression might be involved. Chronic ECS upregulates the expression of
Bdnf and
Creb in the hippocampus, and such upregulation has been shown to mediate antidepressant activity in animal models (
25–
27). Chronic ECS produced chromatin remodelling changes that were very different from those seen after acute ECS, and that were detected at distinct
Bdnf promoter regions. Chronic ECS increased H3 acetylation (instead of H4 acetylation as seen after acute ECS) at
Bdnf promoters 3 and 4, in correlation with increased expression of the corresponding
Bdnf transcripts (Box 2).
Chronic social defeat stress, an animal model of depression (
28) that mimics many symptoms of human depression, also alters chromatin regulation of
Bdnf (
29). Prolonged exposure to an aggressor induces lasting changes in mouse behaviour such as social avoidance, which are reversed by chronic (but not acute) treatment with antidepressants (
28,
29). At a molecular level, chronic defeat stress in mice induces sustained downregulation of the expression of two splice variants of
Bdnf, BdnfIII and
BdnfIV, in the hippocampus (
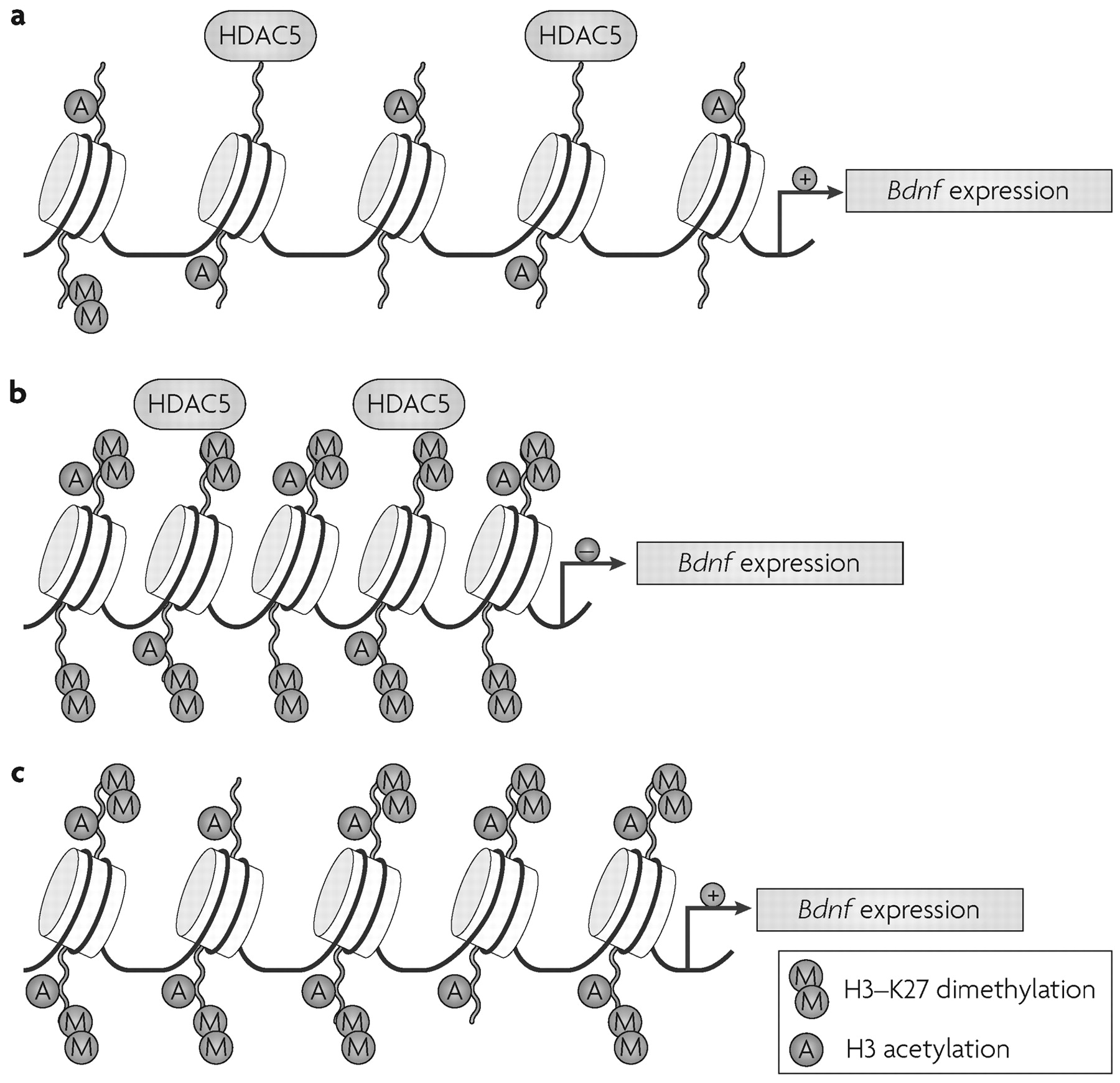
29). These changes are reversed only after chronic treatment with the antidepressant imipramine. Chronic defeat stress induces robust and long-lasting increases in H3-K27 dimethylation, a repressive modification, specifically at the promoters of the downregulated
Bdnf transcripts.
This histone modification was present four weeks after the cessation of defeat stress and was not reversed by antidepressant treatment, indicating that chronic defeat stress imposes a long-lasting marker of repression at these
Bdnf promoters (
Fig. 3). Rather, chronic imipramine seems to reverse repression of the
Bdnf gene by inducing H3 acetylation, as well as H3-K4 methylation—an activating modification (
Fig. 1)—at the same promoters. Direct support for this hypothesis comes from the observation that chronic imipramine downregulates
Hdac5 expression in the hippocampus, but only in animals previously subjected to chronic social defeat. Moreover, viral-mediated overexpression of
Hdac5 in this region prevents imipramine's restoration of
Bdnf levels as well as the drug's antidepressant effects (
29). Further support for this scheme is the observation that systemic administration of sodium butyrate, a nonspecific HDAC inhibitor (see Supplementary information S1 (table)), acts as an antidepressant in models of depression, including social defeat (
29,
30).
The mechanism by which chronic antidepressant treatment upregulates H3-K4 methylation
in vivo is unclear, but a recent
in vitro study may provide some insight. Demethylation of histone H3-K4 is catalysed by BHC110/LSD1, an enzyme that has a close structural homology to monoamine oxidases. Monoamine oxidase inhibitors, which are a class of antidepressants, can increase global levels of H3-K4 methylation and cause transcriptional derepression of specific genes
in vitro (
31). The importance of HMT and HDM inhibition for the action of antidepressants remains unknown, but this can now be tested because distinct HMTs and HDMs act on H3-K4 versus H3-K27. Together, these studies implicate chromatin remodelling in the formation of stable neuronal adaptations (molecular scars) in the hippocampus that might underlie some of the longlasting changes in behaviour which are induced by chronic stress and reversed by chronic antidepressants. The studies also raise the possibility that specific HDAC, HMT and HDM inhibitors might be useful as antidepressant treatments (
Fig. 3).
Stressful events early in life might also leave lasting epigenetic marks on the organism. In rats, some mothers naturally display high levels of nurturing behaviours, such as licking, grooming and arched-back nursing (high-LG-ABN), whereas others have low levels of such behaviours (low-LG-ABN) (
32). Offspring of highLG-ABN mothers are less anxious, have attenuated corticosterone responses to stress and display increased expression of glucocorticoid receptor (GR) mRNA and protein in the hippocampus when compared to pups of low-LG-ABN mothers (
33). The upregulation of
GR mRNA specifically involves the alternatively spliced variant
GR17. The promoter that drives expression of this variant is brain specific, and contains a binding consensus sequence for nerve growth factor inducible factor A (NGFI-A), the expression of which is upregulated in the pups of high-LG-ABN mothers. A study that compared the methylation status at individual CpG sites within the NGFI-A consensus sequence region of the
GR17 promoter between the offspring of high- and low-LG-ABN mothers found that low-LG-ABN pups have increased methylation at these sites (
34). This difference in methylation emerged in the first week of life and persisted into adulthood. As adults, the offspring of mothers with low levels of nurturing behaviour were at a molecular disadvantage—their methylated
GR17 promoter prevented binding of the transcriptional enhancer NGFI-A, effectively disrupting the normal transcriptional regulation of the
GR gene. Although this epigenetic methylation was long-lasting, it could be reversed by infusion of the class I and II HDAC inhibitor trichostatin A (TSA) (see Supplementary information S1 (table)), which increased levels of H3 acetylation, cytosine demethylation and the binding of NGFI-A at the
GR17 promoter in the low-LG-ABN-raised offspring. Interestingly, cross-fostering also reversed the differences in methylation at that site (
34).
DNA methylation has traditionally been viewed as an irreversible epigenetic marker; however, this study indicates that patterns of DNA methylation might be reversible even in adult neurons. Enzymes that mediate DNA demethylation in the brain have yet to be identified. Another important concept illustrated by the reversal of DNA methylation by an HDAC inhibitor is the interaction between histone modifications and DNA methylation. It has been proposed that demethylation might occur through activation of second messenger signals that lead to the recruitment of HATs, which, through increased histone acetylation, would allow DNA demethylases greater access to the
GR promoter (
33). Other examples of rapidly reversible changes in DNA methylation in the adult brain have been reported in learning and memory (
35) (see below).
Together, these studies indicate that several epigenetic modifications, including lasting changes in histone acetylation, histone methylation and DNA methylation, are important in animal models of stress, depression and antidepressant treatment. However, this work has so far focused only on the
Bdnf and
GR genes, and only on the hippocampus. Work is now needed to investigate other brain regions that have been implicated in depression and its treatment (
24), and to investigate the involvement of additional genes in mediating the long-term effects of stress and antidepressant treatments on gene transcription.
Epigenetic mechanisms in addiction
Drugs of abuse control human behaviour by hijacking the brain's natural reward centres, including the mesolimbic dopamine system. This circuit includes dopaminergic neurons of the ventral tegmental area (VTA), which project directly to the nucleus accumbens (NAc)—the ventral portion of the striatum. During the past few decades, much has been learned about the brain's reward pathway and how drugs of abuse usurp its functions (
36,
37), but it is unclear why addictive behaviours persist long after drug abstinence, underlying high rates of relapse. Many studies have identified drug-induced changes in mRNA levels in the VTA, NAc and other relevant brain regions (
38–
43). Some of these changes in gene expression persist even after months of abstinence (
44). The longevity of the changes has driven research into chromatin remodelling as the molecular basis of sustained, even life-long, alterations in gene expression in brain reward regions (
45).
An acute dose of cocaine induces the expression of
c-Fos and
FosB in the striatum, and this induction is associated with transient increases in H4 acetylation within 30 min of drug injection (
13,
14). CBP, with its intrinsic HAT activity, is an important mediator of cocaine-induced acetylation of histones at the
FosB gene, and probably serves this function for other genes as well (
46). Acute cocaine administration also induces H3 phosphoacetylation at the
c-Fos gene promoter only (
14), and this effect requires the protein kinase MSK1, as mentioned above (
13).
Chronic cocaine treatment and self-administration activate and repress many distinct genes compared with acute treatment.
FosB, for example, is induced by both acute and chronic cocaine exposure, but acute exposure produces H4 acetylation whereas chronic treatment causes H3 acetylation (
14). Genes that are selectively induced in the chronic state, such as
Cdk5 and
Bdnf (
44,
47), also seem to be acetylated selectively on H3. Interestingly, cocaine induction of histone acetylation at the
Bdnf promoter builds during a week of withdrawal (
14), and precedes the progressive increase seen in BDNF protein and mRNA levels in this region (
44). A similar switch from H4 to H3 acetylation is seen in the hippocampus after acute versus chronic ECS (
22), indicating that H3 acetylation may represent a common, chromatin-mediated mark of persistently or repeatedly activated genes. Little is known about the catalytic selectivity of HATs and HDACs for the specific acetylated residues on H3 and H4. It would be interesting to determine whether this switch from H4 to H3 acetylation is mediated by the recruitment of distinct HATs or HDACs to regulated genes after acute versus chronic treatment.
Cocaine regulation of histone acetylation at the
Cdk5 and
Bdnf genes highlights the importance of exploring genome-wide chromatin structure—by use of ChIP on chip (
48,
49) or SACO (
50) assays, for example—to identify many other genes, the dysregulation of which at the chromatin level contribute to cocaine addiction. Genome-wide epigenetic approaches have yielded exciting results in the fields of developmental and cancer biology (
48,
49), and efforts to carry out similar studies in addiction are now underway. Preliminary evidence from our laboratory has identified several hundred genes that are significantly hyper- or hypoacetylated after chronic cocaine treatment (
51–
53), and future studies will focus on the role of these chromatin changes in the pathogenesis and maintenance of addiction.
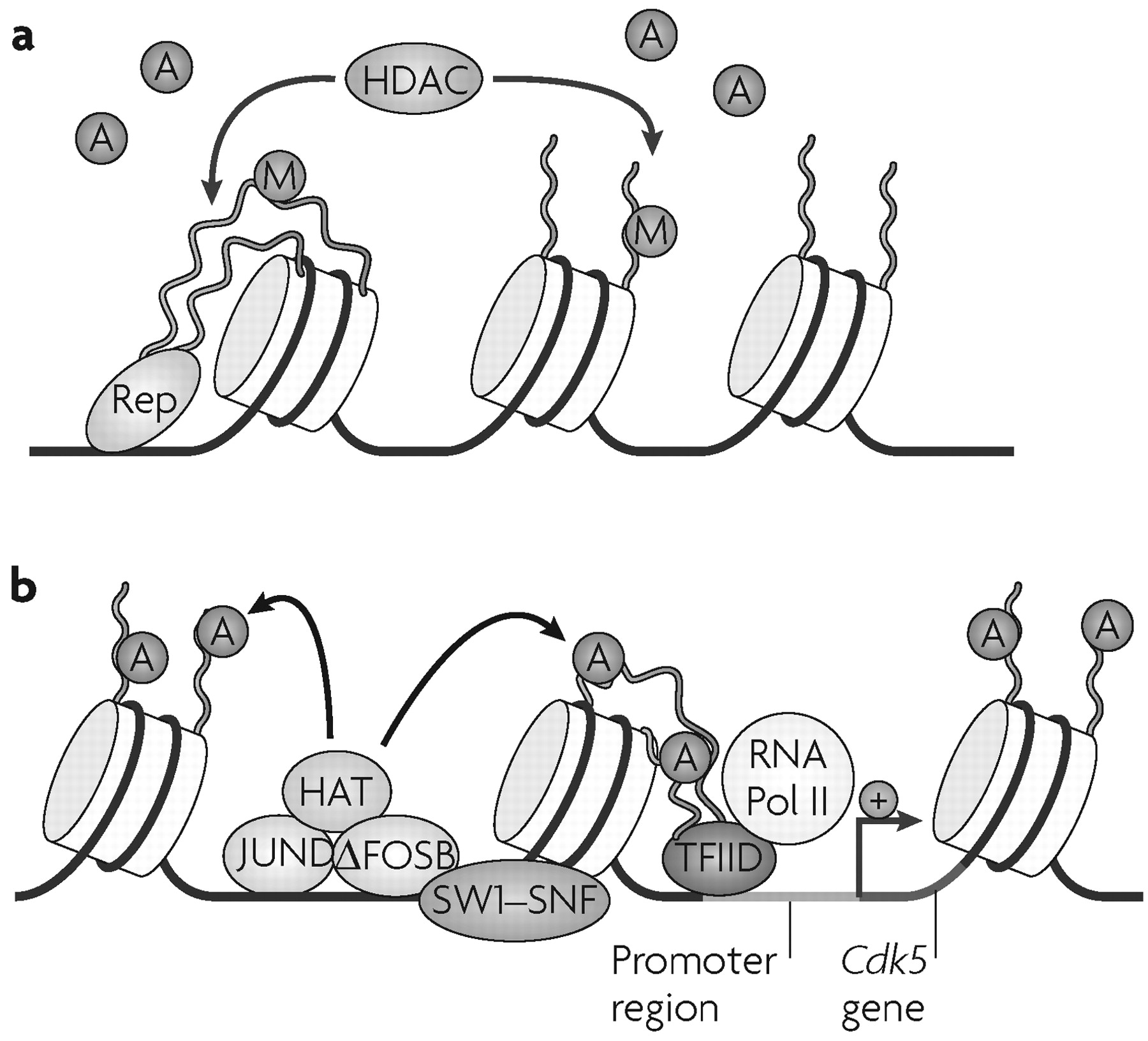
As mentioned earlier, the transcription factor ΔFOSB is implicated in the transition to an addicted state (
3,
4), and accounts for >25% of all changes in steady state mRNA levels induced by cocaine in the NAc (
41). ChIP assays have shown that the induction of one of these mRNAs,
Cdk5, by cocaine represents a direct, activating effect of ΔFOSB on the
Cdk5 gene (
14) (
Fig. 4). By contrast,
Bdnf is not a direct target of ΔFOSB, indicating that a different mechanism is involved in the induction of this gene. Induction of
Cdk5, in turn, partly mediates the effects of chronic cocaine treatment on dendritic remodelling in the NAc (
54). These findings support a model in which the accumulation of ΔFOSB interacts with chromatin remodelling factors at specific promoters to regulate genes that are important for the development and maintenance of addiction (
Fig. 4).
Further studies are needed to identify the enzymes that mediate these cocaine-induced chromatin remodelling events. Histone and DNA modifying complexes are obvious targets. A recent report found that chronic cocaine treatment increases MeCP2 and MBD1 in the adult brain (
55), both of which bind to methylated DNA and aid in transcriptional repression. Several proteins that have been implicated behaviourally in drug addiction are now being rediscovered as histone modifying enzymes. For example, CDK5 phosphorylates an HDAC-interacting protein, which then alters chromatin structure (
56). Another example is the transcriptional regulator named nucleus accumbens 1 (NAC1), which is highly induced by cocaine in the NAc and interacts with HDAC3 and HDAC4 (
57). Possible effects of CDK5 and NAC1 on chromatin structure in animal models of addiction remain unexplored, but these studies might provide mechanistic insight into how drugs of abuse can direct chromatin-modifying enzymes to specific genes. Further work is also needed to examine cocaine regulation of other chromatin-modifying enzymes, and their specific involvement in shaping the addicted state.
The manipulation of chromatin remodelling enzymes by pharmacological inhibition, viral-mediated gene transfer and genetic mutations in mice have shown that cocaine-induced changes in chromatin structure are behaviourally relevant. HDAC inhibitors increase histone acetylation and potentiate cocaine-induced locomotor activity and cocaine reward, as measured by conditioned place preference (
14,
53). Conversely, viral-mediated overexpression of
Hdac4 or
Hdac5 in the NAc had the opposite effect. Consistent with these findings, mice deficient in CBP show reduced cocaine-induced locomotor activity (
46). Together, these findings indicate that the ability of cocaine to increase histone acetylation is important for the behavioural effects of the drug, and set the stage for more in-depth analysis of HDAC inhibitors and related pharmacological and genetic tools in paradigms of cocaine self-administration and relapse.
There are also a few reports of chromatin changes after chronic ethanol administration (
58–
60). An early study used circular dichroism to show that chronic ethanol induced a more open structure to chromatin, consistent with a switch from heterochromatin to euchromatin (
58). Subsequently, changes in histone acetylation and DNA methylation have been reported in alcoholic patients (
59,
60).
Future directions
There is much evidence that epigenetic regulation is involved in neurogenesis, neuronal plasticity and learning and memory, and in disorders such as depression, addiction, schizophrenia and cognitive dysfunction. Changes in histone modifications and DNA methylation have been found both globally and at the promoters of genes that have been implicated in these phenomena. For example, chromatin remodelling at the
Bdnf promoter is associated with neuronal activity, seizures, chronic stress, cocaine addiction and Rett syndrome; at the reelin promoter with a mouse model of schizophrenia; and at the
Cdk5 promoter with drugs of abuse. However, chromatin remodelling is likely to affect many more genes, and it is important to investigate this in both animal models of psychiatric conditions and post-mortem human brain tissue. Coordinated gene expression arrays and ChIP on chip arrays are likely to be helpful in elucidating the promoter gene targets for histone modifications as well as the
in vivo binding sites of transcriptional activators or repressors in relevant brain regions. Such studies will provide a global picture of epigenetic regulation, which is currently lacking. Genome-wide epigenetic approaches have yielded exciting results in the fields of developmental and cancer biology (
48–
50), and efforts to carry out similar studies in addiction and depression are ongoing. Many genes show abnormal levels of histone acetylation or methylation after chronic cocaine administration or stress, and future studies will focus on the influence of these global changes in chromatin on the pathogenesis of addiction and depression, and on identifying the transcription factors that mediate this regulation. It will be necessary to use a system-based biological approach to interpret the data from such studies (
92).
The lack of specific inhibitors for many epigenetic regulators impedes progress in understanding the mechanisms by which chromatin remodelling contributes to psychiatric disorders. As just one example, all available HDAC inhibitors act broadly and inhibit all Class I and Class II HDACs (see Supplementary information S1 (table)). Moreover, certain HDACs (Class III HDACs, termed sirtuins, as well as HDAC6, a Class IIb HDAC) can deacetylate proteins other than histones (for example, tubulin, MEF2, the tumour suppressor p53 and the DNA repair protein Ku70), and these non-histone actions complicate interpretation of the inhibitors' biological effects. Inhibitors that are specific for particular HDACs or other chromatin modifiers would allow us to identify the specific players in the epigenetic regulation of psychiatric phenomena. Such specific inhibitors would also have the potential of being used as new treatment agents for these disorders. In addition, it will be important to generate mouse models that lack particular chromatin-related genes in order to study their molecular and behavioural influences.
Lasting changes in chromatin modifications in animal models often occur only after chronic behavioural manipulations, which try to mimic the long-lasting behavioural changes associated with psychiatric disorders. A better understanding of the mechanisms by which such stable changes are brought about would not only advance our knowledge of the basic neurobiology of these illnesses, but might also provide new therapeutic avenues for disorders such as depression, addiction, schizophrenia and neurodevelopmental illnesses.