I begin this essay with the question of the diagnostic significance of familial aggregation. How much does the tendency for a syndrome to run in families (or be heritable in twin studies) tell us about its underlying diagnostic validity? We then examine the degree to which genetic strategies can inform us about major nosologic conundrums such as the relationship between schizophrenia and bipolar illness. In particular, will application of molecular genetic strategies produce new insights into these old problems? Next, we explore, from a nosologic perspective, the attractions and failures of the Mendelian model for psychiatric disorders. In so doing, we examine how findings in psychiatric genetics can impact on our understanding of the basic nature of psychiatric disorders. Especially, can genetics research tell us whether psychiatric disorders are true entities defined by their underlying nature? Finally, now that we are beginning to identify and replicate susceptibility genes for psychiatric disorders, we explore the nosologic implications for such findings. In particular, to what degree can we anchor our diagnostic concepts on disease genes, even as the basic definition of the nature of a gene is shifting in light of advancing knowledge?
THE LIMITS OF FAMILIAL AGGREGATION OR HERITABILITY AS A MEASURE OF DIAGNOSTIC VALIDITY
Although evidence that psychiatric disorders are inherited (or “run in families”) had been noted by psychiatric clinicians of past generations, the first attempt to use this finding in a formal diagnostic process was taken by Robins and Guze in 1970 (
2). In their seminal paper, they proposed five phases of diagnostic validation, the last of which was titled “family study.” About this fifth phase, they wrote:
Most psychiatric illnesses have been shown to run in families, whether the investigations were designed to study hereditary or environmental causes. Independent of the question of etiology, therefore, the finding of an increased prevalence of the same disorder among the close relatives of the original patients strongly indicates that one is dealing with a valid [diagnostic] entity.
To be useful, a validating criterion must have both high sensitivity (to validate most syndromes that are true disorders) and high specificity (to invalidate most syndromes that are not true disorders). Only in this case can we be confident that syndromes meeting the validating criterion are likely to be true disorders.
Although the criterion of familial aggregation probably has high sensitivity (most true psychiatric disorders run in families), it has poor specificity because lots of things that run in families are not valid diagnostic entities. This point can be illustrated with the following scenario, in which physical rather than mental characteristics are central: A new disorder, “syndrome Z,” is proposed with three diagnostic criteria: 1) height over 6 ft, 2) red hair, and 3) a large nose. A family study of syndrome Z collects 100 affected individuals and 100 control individuals and then examines all first-degree relatives. A substantially higher prevalence of syndrome Z is found in the relatives of the affected individuals than in the relatives of the controls. On this basis, syndrome Z is declared to meet the “family study” validity criterion of Robins and Guze.
Since height, hair color, and nose size all “run in families,” a syndrome constituted of these three traits will, ipso facto, also be familial. The application of the “family study” criterion to syndrome Z will produce a false positive result. Because the preponderance of human psychological and physical traits are familial, such false positive results are likely to be common, undermining the value of the validating criterion of familial aggregation.
In their generally thoughtful book, McHugh and Slavney (
3) made a similar error of inference. They suggested that psychiatric syndromes can be viewed from four perspectives: as diseases, dimensions, behaviors, and life stories. Each of these perspectives, they suggested, is appropriate for certain psychiatric disorders. In discussing whether anxiety is best conceptualized as a dimension or a disease, they wrote:
Anxiety can also be the cardinal feature of attacks of the panic-anxiety state, a psychiatric condition that has been documented as probably a disease by demonstrating its heritability (3, p. 142).
McHugh and Slavney suggested that heritability of a syndrome supports its being considered a disease rather than a disordered behavior or the pathological end of a dimensional process. As with Robin and Guze's criterion for familial aggregation, this claim might have high sensitivity, because most true psychiatric diseases are heritable. However, their claim will have low sensitivity because a large proportion of other human physical, psychological, and behavioral traits are also heritable. In particular, syndromes characterized by disordered behavior (including drug and alcohol abuse, antisocial behavior, and bulimia), symptoms of anxiety, and all major dimensions of personality (several of which are strong risk factors for certain psychiatric syndromes) are all substantially heritable (
4–
9). A decision about whether to call a syndrome a disease requires the considerations of other factors in addition to its degree of heritability.
THE LIMITS OF GENETICS AS A TOOL TO ADDRESS DIAGNOSTIC CONUNDRUMS
Examining disorders one at a time in genetic designs provides nosologic information of limited value. This is not true when two or more disorders or criteria sets are examined. For example, in comparing two approaches toward diagnosing the same disorder, showing that one produces a higher degree of familial aggregation is useful information for the nosologist. Assume we have one well-established disorder–A–and a new disorder–B–that might be closely related. It would be of substantial nosologic interest to determine if, in a family study, disorder B occurs at elevated rates in the relatives of probands with disorder A. If the answer to this question is yes (as, for example, has been seen for schizophrenia and schizotypal personality disorder (
10)), then there is evidence that these two disorders shared familial etiologic factors, which in turn might suggest a nosologic relationship. If the answer is no, diagnostic independence would be favored.
These same questions can also be asked, with greater conceptual clarity, by twin or adoption designs that can isolate genetic from familial-environmental effects. Twin studies—which assess all genetic effects together at the aggregate level—have in particular proven useful in determining the overall genetic relationship between different disorders, quantified in the statistic termed the genetic correlation. For example, the genetic correlation is very high for major depression and generalized anxiety disorder (
11) but lower for major depression and animal phobia (
12) and alcohol dependence and pathological gambling (
13).
Although such results can
inform nosologists' decisions, they cannot, in and of themselves, answer the fundamental nosologic questions. This is true for at least two reasons. First, taking twin studies as an example, these studies alone cannot address the issue how high a genetic correlation has to be to consider two syndromes to be subtypes of a single disorder or how low a genetic correlation has to be to consider the two syndromes to be independent diagnostic entities. Second, such studies alone cannot answer questions such as What should nosologists do if two disorders have closely related genetic risk factors but distinct environmental risk factors or are genetically distinct but respond to the same kinds of treatment? and Should genetic risk factors be given highest priority in such nosologic decisions? Such questions cannot be addressed by purely empirical means but require judgments about the relative importance, in a particular nosologic decision, of different potential validators (
14).
However, family, twin, and adoption studies are now not the only approach in psychiatric genetics that can be applied to diagnostic conundrums. Linkage and association studies can provide information, respectively, about whether genomic regions or specific genes influence risk for more than one disorder. Will these newer methods prove of greater value to the psychiatric nosologist? Might it be possible that just as molecular genetics has been used in biology to help define species, it might be similarly used to define psychiatric disorders?
As an example of this approach, Berrettini (
15) reviewed evidence from linkage studies suggesting regions with linkage to both schizophrenia and bipolar illness. He wrote:
Review of these data indicates that there are five genomic regions that may represent shared genetic susceptibility of BPD [bipolar disorder] and SZ [schizophrenia]. As the genes underlying these confirmed linkages are identified, the current nosology must be changed to reflect the new knowledge concerning the shared etiologies of BPD and SZ.
There are at least two potential caveats to these claims:
1.
different genes under the same linkage peak could influence liability to schizophrenia and bipolar illness, and
2.
these regions of “joint” linkage might arise by chance, given the large number of chromosomal regions putatively linked to each disorder. However, some evidence, which is still quite preliminary, has suggested that individual candidate genes may be associated with both schizophrenia and bipolar illness (e.g., see references
16–
18). In one such study, using a modest sample size, Hodgkinson et al. (
16) found several alleles at single-nucleotide polymorphisms in the
DISC1 gene that were significantly associated with schizophrenia (with odds ratios of 1.2 to 1.3) and other alleles in the same gene that were significantly associated with bipolar illness (with odds ratios varying from 1.1 to 1.2). What impact should it have on our nosology if one or more genes are shown definitively to be associated with both schizophrenia and bipolar illness?
The liability to multifactorial disorders such as schizophrenia and bipolar illness are almost certainly influenced by a large number of genes (
19,
20). For the sake of argument, assume that each of these two disorders is influenced by variants at 20 different genes. What would it mean if verified findings emerged that one, two, or even five susceptibility genes were shared between these two disorders? At what point should we alter our nosology on the basis of such findings?
General medicine contains many examples of distinct diagnostic categories that share genetic risk factors. For example, genes that predispose an individual to essential hypertension (
21) will increase the liability to hemorrhagic stroke, myocardial infarction, and hypertensive cardiomyopathy. Mutations in the oncogene
BRCA1 increase risk for cancer of the breast, cervix, uterus, pancreas, fallopian tube, stomach, colon, and prostate (
22). Yet, this evidence for “common genes” has not been used to support changes in the classification of these disorders.
Finding a small number of genes that influence susceptibility to two multifactorial disorders is not likely to provide definitive information for nosologists. Indeed, from a nosologic perspective, this information differs little from finding a modest genetic correlation between two disorders in twin studies.
A different situation would emerge if, as we identify susceptibility genes, we found that all or nearly all the genes that predisposed to disorder A also predisposed to disorder B. Such a finding, which would represent a confirmation at a biological level of results from family studies (that is, high levels of coaggregation) or twin studies (a high genetic correlation), would provide further evidence that the two disorders were closely related.
To illustrate a likely pattern of results that will emerge for pairs of related psychiatric disorders, it is informative to review what has been learned about the genetic relationship between the two major forms of inflammatory bowel disease—Crohn's disease and ulcerative colitis. Most, but not all, family studies have indicated modest levels of coaggregation, with better evidence that rates of ulcerative colitis are increased in relatives of Crohn's disease probands than vice versa (
23,
24). Twin studies have suggested that both disorders are heritable. Monozygotic twin pairs where one twin has Crohn's disease and the other ulcerative colitis are, however, rare (
23). In a recent meta-analysis of 10 linkage studies of inflammatory bowel disease, suggestive evidence for linkage was found in six regions for Crohn's disease and in only one region for ulcerative colitis (
25). However, the single region of tentative linkage for ulcerative colitis (2q) was one of the six found for Crohn's disease. Association studies have shown that variants in the best replicated susceptibility gene for Crohn's disease (
CARD15) do not influence risk for ulcerative colitis (
24). A high-risk haplotype identified in chromosome 5q31–33 predisposed to risk for Crohn's disease but not ulcerative colitis. However, variants in the human leukocyte antigen region have been found that increase risk for both disorders (
24).
What should a nosologist conclude from these results about the relationship between Crohn's disease and ulcerative colitis? Some genetic risk factors are shared, and others appear distinct. Would this pattern of results—which may be common for moderately related complex syndromes in biomedicine—lead easily to a clear decision about the nosologic relationship between the two disorders? It seems unlikely that molecular genetics will bring the same clarity to the classification of complex diseases in medicine as it has for species in biology.
It is worth asking at this point whether we are at risk for adopting too “gene-centered” a view of psychiatry—of making too much nosologically of the modest effect sizes we are finding for individual genes. For example, severe sexual abuse in an epidemiologic sample of women increased the risk both for major depression and for drug abuse with odds ratios of 3.14 and 5.70, respectively (
26). Although these figures are much greater than that seen in the studies showing association of the same gene with schizophrenia and bipolar illness, such findings have not lead to suggestions to modify our nosology to reflect shared etiologies of depression and substance abuse.
THE LIMITS OF GENE DISCOVERY FOR PSYCHIATRIC NOSOLOGY, OR “ONCE WE FIND THE GENES…”
At a conference in 2004, the following conversation between two psychiatric genetics researchers was overheard:
Researcher 1: Part of what has made advances in our field so difficult is the probable heterogeneity of the disorders that we study. It is hard to believe that schizophrenia, alcoholism, or depression are really one disorder. But our clinical tools have not been very successful at pulling these disorders apart into purer, more etiologically homogeneous entities.
Researcher 2: Yes, I agree completely. But once we find the genes for these disorders, then things will start to change. We will finally be able to provide a firm scientific foundation for psychiatric diagnoses. We can stop having these endless debates and be able to solve all these problems once and for all.
By 2004, it had become clear to everyone in the field that no “Mendelian-like” genes for psychiatric disorders were likely to be found. Nonetheless, there is continued hope that advances in psychiatric genetics and particularly the identification of individual susceptibility genes will alter, in fundamental ways, our approach to psychiatric diagnosis. If we are able to find a “gene for” a particular psychiatric disorder, then we can work our way back up and—as predicted by the EGM—ground our diagnostic category on the firm foundation of a gene.
Categorical gene models and the problem of small effect size
One of the hopes expressed by these researchers is that new discoveries in psychiatric genetics will permit us to define the boundaries of psychiatric syndromes. They have expressed a second implicit expectation for the nosologic impact of psychiatric genetics research—that it will support categorical definitions of illness. A categorical view of psychiatric illness—that these disorders are discrete entities with distinct boundaries—can be contrasted with the perspective that these disorders are pathological ends of functional continua.
Advocates of a categorical perspective on psychiatric disorders suggest that they are discrete entities that are similar to biological species, such as whales and cows, or to man-made objects such as shirts and pants. Such entities have distinct boundaries. It is clear what is inside and what is outside. With categorical entities, the task of the nosologist becomes finding these boundaries or, as is oft said, “carving nature at its joints.”
Classical Mendelian disorders appear to be such categories. Clinically, these diseases appear to be discrete. In families with multiple affected individuals, there are typically no “spectrum” cases; individuals are either affected or unaffected. Perhaps genetic discoveries would uncover such clear forms of psychiatric illness.
Categorical models of disease, by definition, require discrete boundaries or what has been termed “points of rarity.” For genes to be useful in defining categorical disease entities, the etiologic effect of the gene must be large enough that it produces such a “point of rarity” between those who possess and those who lack the disease gene (
35). Classically, this would be represented as bimodality in a distribution of liability—the joint that is to be carved. To successfully ground a categorical diagnostic system in pathogenic genes, the genes need to affect liability strongly enough that their impact is detectable above the background effect of other risk factors.
As reviewed earlier, prior evidence from linkage studies of psychiatric disorders and animal behavior genetics do not provide encouraging news for the EGM. The effect sizes of genes found in these studies have typically been small. In the last decade, individual susceptibility genes have been tentatively identified for psychiatric disorders. Therefore, we can examine the magnitude of their effect more directly. A review of positive meta-analyses of functional candidate genes for psychiatric disorders found odds ratios ranging from 1.07 to 1.57, with a median of ∼1.30 (
36). (An odds ratio is the risk for a disorder given the presence of a risk factor—here a particular gene—divided by the risk for that disorder in the absence of exposure to the risk factor.) In schizophrenia, replicated evidence is now emerging for several genes that have been localized under linkage peaks (
37), in particular dysbindin 1 and neuregulin 1. A recent review of dysbindin studies suggested that the odds ratio of variants in the gene and risk for schizophrenia average around 1.50 (
38). For neuregulin 1, two recent replications reported odds ratios of 1.25 and 1.80 (
37).
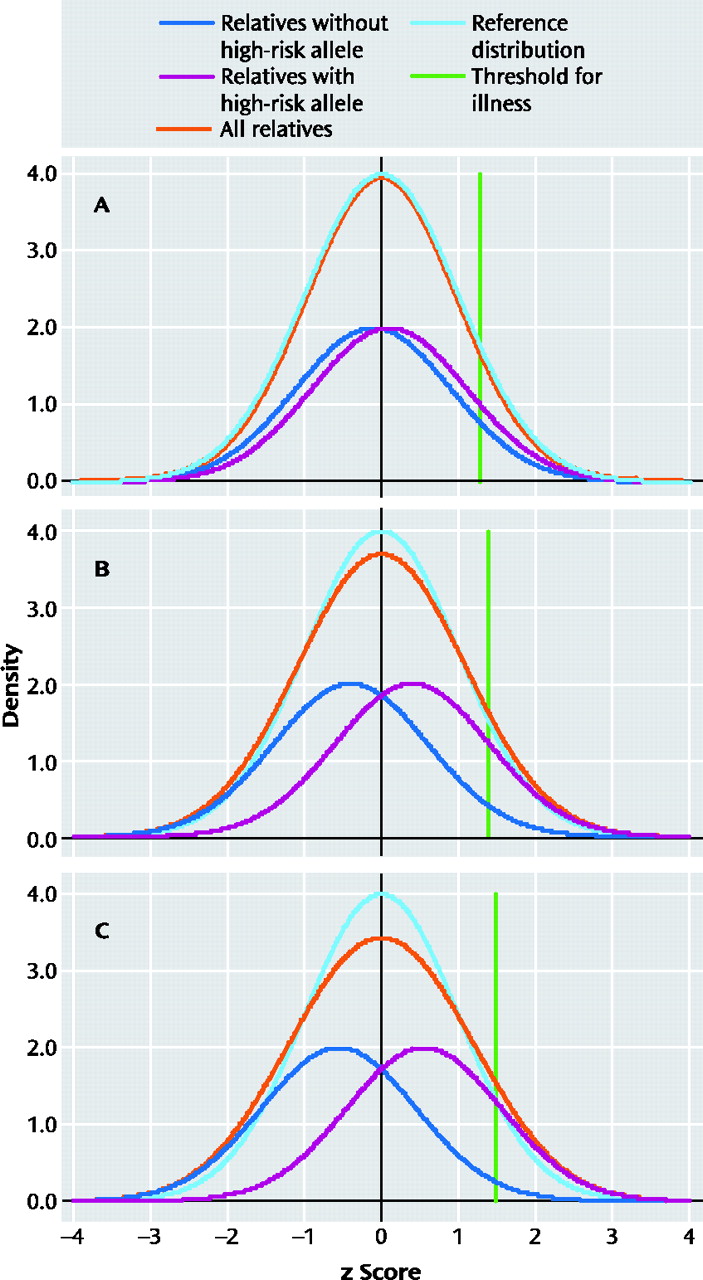
Figure 1 displays the liability distributions in a putative sample of first-degree relatives of individuals with schizophrenia, one-half of whom possess a high-risk copy (more technically allele) of a gene for schizophrenia. (The term “liability” here reflects the individual's level of risk for the illness, with risk increasing as you move from left to right in each panel in
Figure 1.)
Using plausible parameters (see
Figure 1 legend for further details), we have varied the magnitude of risk conveyed by the gene to produce odds ratios for the relationship between the high-risk allele and schizophrenia of 1.5, 5, and 10 in
Figure 1 panels A, B, and C, respectively. Each panel presents four different distributions of liability. The dark blue line reflects the liability distribution of relatives without the high-risk allele. The purple line reflects the liability distribution of relatives with the high-risk allele. The turquoise line reflects the “reference” liability distribution that would be seen if there were no individual genes of detectable effect and only background genetic and environmental variation that would be predicted to take the shape of a normal distribution. The orange line, which is the most important one, reflects the liability distribution of the population of all relatives and is simply the sum of the blue and purple line. In addition, the green line represents the cutoff point for illness. Individuals with liability above that threshold will develop illness.
The thought experiment we are here conducting is as follows: If we could measure liability directly in these relatives (although we would not know their individual genotype), could we cut cleanly (at nature's joint) between those at high risk (depicted by the purple line) and those at low risk (depicted by the blue line)? That is, in the total population distribution (depicted by the orange line), do we see a clear point of rarity separating the two groups? For a gene with an odds ratio of 1.5 (
Figure 1, panel A), there is virtually no deviation in the population distribution from that predicted by a single normal distribution. Even with odds ratios of 5 or 10 (
Figure 1, panels B and C, respectively), all that is observed is a slight flattening of the distribution with no evidence for a point of rarity at which to divide those carrying and not carrying the high-risk allele.
Effect sizes are the range of those seen with genes that impact on risk for psychiatric disorder are too small to produce, on their own, syndromes with discrete boundaries.