The physiologic relevance of the electroencephalogram (EEG) as a reflection of neural activity
1 and the evidence of abnormal neural activity in cocaine dependence
2–4 suggest the possibility that the EEG might reflect abnormal neurobiology in cocaine dependence. The hypothesis presented in this article links abnormal EEG findings observed in cocaine dependence
2,5–10 to stimulant sensitization. Although such a discussion is necessarily speculative, it might ultimately prove to be useful given the potential advantages of the EEG as a means of studying cocaine dependence in humans. The EEG is noninvasive and inexpensive, and quantitative analysis of the EEG (qEEG) is based on digital technology that is constantly growing in analytic power and accessibility. Clinically, the EEG is sensitive to psychiatric conditions such as depression or attention-deficit/hyperactivity disorder (ADHD) that are often comorbid with cocaine dependence.
11,12 Pretreatment qEEG has shown a correlation with subsequent psychotropic drug response in patients with a variety of psychiatric disorders evaluated prospectively.
11 Using the EEG to gain access to a neural correlate of sensitization could possibly be useful in the investigation of its relationship to the clinical syndrome of cocaine dependence, and ultimately in the development or selection of medications according to a hypothesis of stimulant sensitization.
The more consistent neurochemical findings suggestive of persistent neuroadaptive changes in stimulant sensitization include increased DA efflux in the nucleus accumbens (NAc) in response to a cocaine or amphetamine challenge
14,15,18,19 and enhancement of signal transduction through DA D
1 receptors
15,20–23 and inhibition of NAc neurons.
24–26 The latter two phenomena are of particular interest with regard to sensitization and the EEG because they can manifest on a tonic, continual basis, and therefore they occur on a temporal scale consistent with the spontaneous resting EEG. The robust consistency of the phenomenon of stimulant sensitization in animal experimental paradigms and the apparent involvement of neural systems implicated in reward and motivation have led to significant interest in the possible role of sensitization in the clinical syndrome of cocaine dependence.
13,14,27,28 It has been suggested that a sign of sensitization, namely increased DA efflux in the NAc, is a correlate of salience and motivational relevance, and that its augmentation over time in response to cocaine or cocaine-related cues with sensitization reflects the increasing attentional and motivational focus on cocaine and cocaine-related stimuli.
28–30In the following discussion, the literature on the neurobiology of cocaine sensitization is reviewed in the context of the generation and dopaminergic modulation of the EEG, and the hypothesis is presented that the EEG power spectral profile observed in cocaine withdrawal reflects changes in DA neurotransmission that are associated with stimulant sensitization.
COCAINE AND THE EEG
The reported acute and chronic qEEG effects of cocaine in animals and humans generally involve some combination of enhanced fast activity, diminished slow activity, and broad reductions in EEG amplitudes across the entire power frequency spectrum. These are EEG states to which terms such as
desynchronization or
arousal31 have been applied. That common attributes have been observed between EEG states following acute and chronic exposure is consistent with the conceptualization of sensitization as a “proponent” process.
32 In a proponent process, similarities exist between the state following acute drug administration and the state of withdrawal following chronic drug administration. In the case of cocaine, specific phenomena with observed consistency between the acute and chronic states include enhanced transmission through DA D
1 receptors and inhibition of NAc neurons.
15The effects of acute and chronic exposure may be viewed as generally consistent with the transition from global to local EEG modes.
33 Global EEG modes tend to originate from sources with a relatively widely dispersed topographic distribution and have high amplitudes and low frequencies. Activity in the “slow” bandwidths delta and theta occurring in normal awake humans and the eyes-closed occipital alpha rhythm are examples of global EEG modes. Functionally, global EEG modes are thought to serve the purpose of gating input or motor output and coordinating activity across widely distributed cortical regions. The apparent gating of sensory input in the visual cortex by the occipital alpha rhythm is an example of the gating function attributed to a global EEG mode. Global modes might also serve the purpose of facilitating coordinated activity between relatively widely distributed cortical expanses, such as the putative role of the theta rhythm in enhancing transmission between temporal allocortex and frontal neocortex.
34,35 Local EEG modes are higher in frequency, lower in amplitude, and distributed over a more limited topographic area. Functionally, local EEG modes tend to correspond to tasks that involve attending to incoming stimuli or outputting motor action. An example of a local EEG mode would be the appearance of beta frequency activity over visual cortex during a target detection task, or over contralateral motor cortex during a manual movement task.
31Acute administration of cocaine in rats reportedly results in diminished slow EEG power and overall amplitudes and increased beta frequency power.
36–39 These EEG effects appear to be mediated by DA to an important extent, as evidenced by their temporal correspondence to DA levels in the prefrontal cortex and their diminution by D
1 antagonist.
36,37 Several findings are consistent with a role of increased transduction through D
1 receptors in sensitization. Reduced global EEG amplitudes have been observed with the administration of either cocaine (10 mg/kg) or a selective D
1 agonist
38,39 and in the spontaneous EEG in drug-free rats previously exposed to cocaine according to a schedule designed to produce sensitization.
40 Cocaine-sensitized rats also reportedly show a greater relative decrement of EEG power in the delta and theta bands in response to an acutely administered dose of cocaine.
40 The possibility that the greater reduction in slow activity in response to acute cocaine in sensitized animals might reflect increased DA signal transduction receives support from an animal study on rats involving ventral tegmental area (VTA) stimulation. Leung and Yim
41 reported on a delta frequency rhythm recorded directly from the NAc, which they termed
accumbens delta, that was desynchronized by electrical stimulation of the VTA; this effect was blocked by haloperidol.
In humans, acute administration of cocaine is most commonly reported to enhance beta frequency activity.
42–44 As has been reported for animals,
40 the acute effects of cocaine on slow EEG activity in humans may differ as a function of prior exposure to cocaine. Relatively inexperienced intranasal cocaine users challenged with intravenous (IV) cocaine have reportedly evidenced a transient delta increase in addition to the expected, and more persistent, beta increase in the first 5 minutes following the injection,
42 which coincides with the expected time of peak euphoria.
45 More experienced IV users tested in a later study did not evidence a delta increase.
44 The transient enhancement of delta activity in relatively naive users, but not in chronic users, could represent a neuroadaptation due to chronic use, possibly sensitization, resulting in a progressive diminution of activity in global EEG modes.
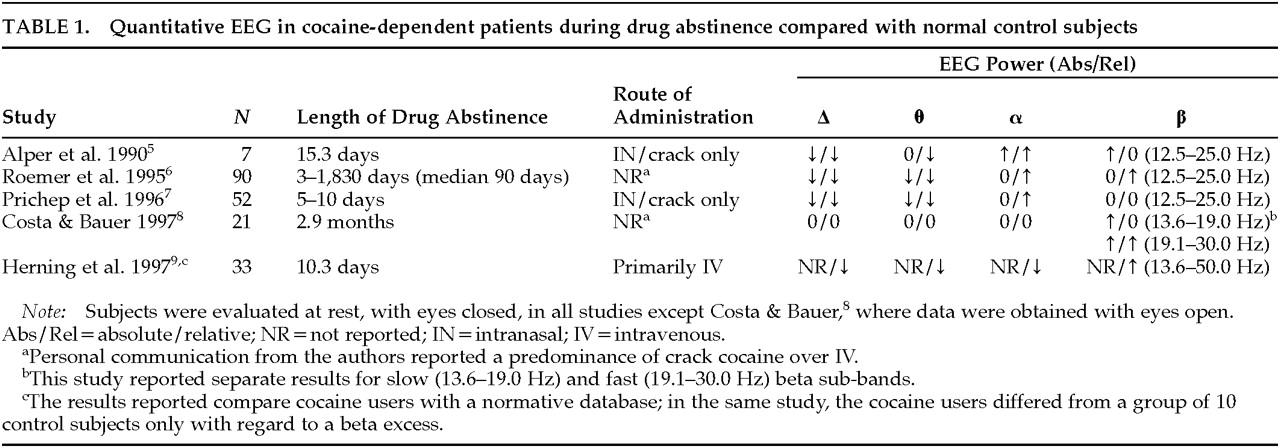
Five studies, summarized in
Table 1, have reported on the qEEG in drug-free subjects with prior histories of chronic cocaine use compared with normal control subjects.
5–9 In the four of these studies in which reduced delta and theta power were noted relative to values derived from normative databases,
5–7,9 visual inspection and automatic detection with exclusion of artifactual epochs were used to control for EEG artifact. The single study that did not note a slow wave deficit
8 relied on a mathematical algorithm to reduce the effect of ocular artifact without the actual exclusion of any artifactual epochs, a methodologic factor that might have been particularly likely to have affected delta frequency findings; also, the lack of a normative database and inclusion of eyes-open data might have limited sensitivity. In four of the studies listed in
Table 1, a relative or absolute beta increase was seen.
5,6,8,9 Discrepant findings regarding beta activity may have been due to electrophysiological heterogeneity between subgroups of subjects
7 and differences related to route of administration. Use of cocaine by the IV route is reportedly associated with greater beta power than cocaine smoking.
46 The study that reported no overall beta excess in crack cocaine users rigorously excluded subjects with a history of any IV drug use
7 and found beta excess only in an electrophysiologic subgroup, defined by cluster analysis of EEG variables, that was characterized clinically by early dropout from treatment.
10 Alpha relative power is reportedly increased in the studies that found an absolute deficit of slow activity,
5–7 and the finding apparently relates more to the slow wave deficit than to an absolute alpha excess. Only one study,
5 which was a pilot sample of 7 subjects, found an absolute alpha power elevation; this finding might have been attributable to the relatively high depressive comorbidity in that sample in view of reports of increased alpha power in depression.
8,11These results in human subjects or animals chronically or acutely exposed to cocaine appear to share the attribute of bias toward the transition from global to local EEG modes, which might be manifested as a diminution of slow or an enhancement of fast EEG activity. Overall, there appears to be significant agreement among the published studies of drug-abstinent, cocaine-dependent humans in finding a slow wave deficit, along with a beta excess in at least a subgroup of subjects. The apparent similarity of the acute and chronic states is consistent with the conceptualization of cocaine sensitization as a proponent process.
32 In the discussion that follows, the hypothesis is presented that cocaine sensitization results in a transition from global to local EEG modes in the frontal cortical terminal fields of the mesotelencephalic DA projection, and that this transition is reflected by diminished delta, a global forebrain EEG mode.
The observation of an association of beta EEG power, a local EEG mode, with treatment failure
10 could logically be related to a sensitization hypothesis. Increased frontal beta activity has also been reported to correlate prospectively with treatment failure in two independent samples of subjects with alcohol dependence.
47,48 In view of the observation that VTA stimulation in animals enhances beta power and diminishes delta power,
41,49 the relative enhancement of beta EEG power in treatment dropouts is possibly consistent with hypotheses linking substance dependence and neural sensitization in the mesotelencephalic DA system.
14,15,18The discussion that follows, however, will focus on the finding of
decreased delta EEG power as a possible correlate of cocaine sensitization. In our studies on crack cocaine dependence,
2,5,7,10 as well as that of Roemer et al.,
6 the delta deficit has been the most statistically abnormal finding and appears to be generally shared across subgroups of patients. A delta deficit has not generally been reported in conditions that could potentially confound EEG results in cocaine dependence, such as alcohol or cannabis dependence or human immunodeficiency virus infection.
11,50–52 A beta excess, on the other hand, is a frequently reported feature in alcohol dependence.
8,11 The abnormal delta findings in cocaine dependence persist for at least 6 months,
2 which is a time frame consistent with the persistence of neural sensitization.
14 We have not been able to determine the persistence of the beta excess that we have seen in the subgroup of subjects who prematurely depart from treatment, since these subjects were not available for retesting after an extended drug-free interval. The discussion that follows will therefore focus on the possible significance of the observation of reduced delta EEG power in cocaine withdrawal as a possible correlate of cocaine sensitization.
FUNCTIONAL SIGNIFICANCE OF THE AWAKE DELTA EEG RHYTHM
The human brain generates delta frequency EEG activity in a variety of neurological and behavioral states, and it is important to specify that the delta EEG power of interest in this discussion is that observed in awake, neurologically normal adults. Delta power can comprise more than 20% of total EEG power in frontal leads in young adults in multiply replicated normative studies.
53–58 The conventional EEG literature emphasizes pathologically excessive delta as a consequence of cortical deafferentation or subcortical inflammatory processes
34 but provides little discussion of the significance of a delta deficit. The term
delta is used herein to refer to the delta EEG rhythm that is observed in neurologically normal, awake adults; it is not the delta seen in sleep, in infancy, or in the presence of neurologic pathology. There is substantial evidence that delta can appear as a correlate of complex cognitive processes. Delta EEG power has been observed to be increased in normal subjects performing calculations,
59,60 reaction time tasks,
61 abstract thought,
62 P300 paradigms,
63,64 and a delayed match-from-sample paradigm.
65Delta EEG activity is an example of a global neocortical processing mode. Global EEG processing modes involve low-frequency EEG activity spanning relatively large cortical regions, as opposed to local modes involving high-frequency EEG activity distributed over smaller cortical areas.
33 Global modes have been hypothesized to serve the purpose of
integration across diverse cortical sites by synchronizing coherent activity and phase coupling across widely spatially distributed neural assemblies. The observation of increased delta EEG power in tasks spanning processes such as working memory or abstraction
59–65 is consistent with a role of delta in the process of integration of activity across association cortex. Global modes might also serve the function of gating extraneous stimuli from gaining access to global assemblies of coherent EEG activity.
1,66 A
gating function has been suggested for delta power in the functional disconnection of the cortex from extraneous thalamic inputs, analogous to the role postulated for alpha in quiescent states of visual sensory cortex.
67 This functional cortical deafferentation has been hypothesized to serve the process of selective attention to information represented in association cortex during states of “internal concentration.”
59,60 Delta in normal awake humans can apparently be a correlate of the process of allocating attention and limiting access of extraneous stimuli during states in which the cortex is processing its own output. Therefore, a delta deficit could conceivably relate to vulnerability toward the preemption of attention by craving and drug-related cues in cocaine dependence.
With regard to the functional role of delta power, it is interesting to note that the other group in our database that most consistently appears to have a delta deficit is subjects with ADHD.
12 The apparently shared feature of a delta deficit in cocaine dependence and ADHD coexists with other evidence of a relationship between the two disorders, which includes a high comorbid incidence, a familial association between cocaine dependence and ADHD,
68 and the common general behavioral features of impulsivity and disturbed regulation of attention.
There is apparently a significant commonality between normal awake delta EEG activity and the P300. The P300 is an evoked response to stimuli that are unexpected, infrequent, or motivationally relevant. In a typical P300 paradigm, a subject is asked to respond to a relatively infrequent target stimulus, as opposed to a more common nontarget stimulus. The amplitude of the P300 increases with the motivational relevance of the task or the salience of the target stimulus, and P300 amplitude reflects the allocation of attentional resources. The psychological processes spanned by P300 paradigms include working memory, motivation, attention, and the appreciation of context.
69 P300 amplitude has been observed to correlate positively with delta EEG power.
64,70,71 Delta EEG activity is selectively enhanced in epochs immediately following target stimuli presented in the context of a P300 paradigm, but it is not enhanced when the same stimuli are presented in a context that does not elicit a P300 response.
63,64 The increased cortical resonance or frequency-specific enhancement of EEG in the delta range during
cognitive and
attentional processes suggests that delta is a correlate of these processes.
POSSIBLE FUNCTIONAL SIGNIFICANCE OF REDUCED DELTA EEG POWER IN COCAINE DEPENDENCE
Reduced delta activity can be viewed as diminished resonance in the delta bandwidth. Resonance is the selective enhancement of signal transmission at specific frequencies. The delta bandwidth is an important global neocortical EEG mode that is apparently involved in the facilitation of neural signal within the neocortex and between the neocortex and subcortical structures. Evidence that delta is a neocortical resonant mode includes the observation of an association of delta frequency EEG activity with partial seizures of neocortical origin, as opposed to the association of theta range activity with allocortical foci.
104 Resonance, to a significant extent, is determined by the frequency of unit activity in the underlying resonant neural structure.
1,34,105 Delta-range unit activity has been observed in neocortical pyramidal neurons that are the putative generator of the surface EEG.
91,93–95 Delta-range unit activity has also been recorded from subcortical structures known to play a prominent role in reward and motivation, including the NAc,
41 the VP,
106 and DA neurons in the VTA.
14 DA-related behaviors are selectively enhanced by stimulation of DA cell axons at 2 to 3 Hz.
14 Delta, a neocortical resonant mode, is a frequency bandwidth over which descending neocortical influences might relatively selectively modulate subcortical DA neurotransmission in structures relevant to reward-mediated behavior.
The functional consequence of the relative diminution of a neocortical resonant mode could involve a relative loss of neocortical modulating influence on subcortical neural transmission. Theta is a resonant mode of allocortex, as evidenced by theta frequency single-unit activity in allocortex
35,107 and the apparent enhancement of neural signal transmission between allocortex and neocortex in the theta frequency bandwidth.
34,35 Although theta is also apparently reduced in cocaine withdrawal, the extent of the reduction of delta is greater.
5–7 The greater reduction in delta, relative to theta, would be expected to correspond to a relatively greater reduction in neocortical versus allocortical influence on subcortical neural transmission. Grace
14 similarly hypothesizes a relative reduction in descending neocortical versus allocortical influence on the basis of changes in the regulation of subcortical DA neurotransmission in stimulant sensitization. As pointed out by Grace,
14 the behavioral correlates of greater allocortical versus neocortical influence on subcortical activity would be expected to include features such as impulsivity, disinhibition, and affective lability that are often evident in the clinical syndrome of cocaine dependence.
28CONCLUSIONS
I have presented the argument that the abnormal power spectral profile observed in cocaine withdrawal is due to cocaine sensitization. Ultimately, for this conclusion to prove to be interesting, sensitization must be confirmed to relate importantly to the clinical syndrome of cocaine dependence, and the EEG must be found to reflect the process of sensitization with a reduction in forebrain global EEG activity. Direct evidence for sensitization and its relationship to addiction in humans would involve the finding of a progressive increase of a sign of sensitized DA transmission, such as increased DA efflux in the NAc, as attention and behavior become more exclusively focused on cocaine and its related cues. There are obvious ethical and practical limits to obtaining such information on the induction of sensitization in humans as they become dependent on cocaine. However, animal work correlating the progressive increase in signs of sensitization with cocaine-dependent behavior and EEG transition from global to local modes could clarify the significance of the apparent loss of global, and enhancement of local, EEG activity in cocaine-dependent humans.
A major difficulty in studying humans with histories of chronic cocaine use appears to be a restriction of the dynamic range of the expression of sensitization in chronic users, due to the prior induction of maximal degrees of sensitization. This effect has been evident in human studies of behavioral sensitization to cocaine or amphetamine. Human chronic cocaine users reportedly do not clearly evidence a sensitized behavioral response to the second administration of a dose of stimulant relative to the first.
108,109 However, stimulant-naive subjects do evidence a sensitized behavioral response to the second of two amphetamine doses.
16,17 It is apparently difficult to demonstrate the induction of sensitization with an interval change between two stimulant doses in chronic users because they may already have attained maximal degrees of expression of sensitization that no longer increment strongly with successive doses.
110 Nonetheless, on the basis of evidence presented here, it is suggested that the EEG may reflect the expression of sensitization in chronic cocaine users, and that EEG might provide a noninvasive, accessible approach to investigating the possible relationship of the expression of sensitization to the clinical syndrome of cocaine dependence in human subjects.