Adrenoleukodystrophy (ALD) is an X-linked peroxisomal disorder in which very long chain fatty acids (VLCFA), defined as those having greater than 22 carbon chains, accumulate within cells as a result of defective beta-oxidation within the peroxisome.
1,2 Although this disorder was initially thought to occur only in childhood, it is now apparent that it can occur over a wide age spectrum with considerable clinical heterogeneity. The frequency of the disorder is essentially unknown, although an incidence of 1:20,000 has been cited.
1The classic or childhood-onset form is a rapidly progressive neurodegenerative disease that begins between the ages of 3 and 8 and is characterized by symptoms of attention deficit disorder, followed by intellectual, behavioral, and neurological deterioration. The illness progresses rapidly to a vegetative state within 2 years and death soon after.
3–6 An adolescent-onset form, which is less severe, may present with primary adrenal insufficiency, neurological dysfunction, or psychiatric symptoms. Death usually occurs within 1 to 2 years of cerebral involvement.
3,4,6 Unfortunately, the designation
adrenoleukodystrophy has also been applied to an autosomal recessive form that is associated with dysmorphism and is fundamentally different from X-linked ALD.
More recently, adult-onset ALD has been described.
1,2 Afflicted individuals may present with any combination of adrenal, gonadal, neurological, or psychiatric disorders. The majority will have neurological involvement at some point during the course of their illness.
4,7 Although the most common presenting features include abnormalities of gait and evidence of upper motor neuron involvement, mania and psychosis can occur first.
8–16 The age of onset can be difficult to determine in the adult variant. Often patients have milder symptoms that are not attributed to the disease, except in retrospect. Many cases of “adult onset” therefore involve symptoms that have been present since childhood or adolescence but did not come to medical attention until much later. In the case of psychiatric symptomatology, it is often difficult to precisely identify the onset of personality or mood changes; if noticed, they are often attributed to other causes. We review the genetics and pathophysiology of the disease and summarize a literature of 34 cases of ALD, describing the neuropsychiatric symptoms and treatment options.
GENETICS OF ALD
The ALD gene has been mapped to X-q28. It contains 10 exons, spans 20 kb of genomic DNA, and codes for a protein called the ALD protein that has been localized to the peroxisomal membrane.
17,18 Although the gene was expected to code for VLCFA CoA synthetase, the enzyme involved in the first step of VLCFA degradation within the peroxisome, it has recently been shown to code for a protein that is a member of the ABC transporter superfamily.
19 This transporter forms a “pathway”
19 through which the enzyme VLCFA CoA synthetase can move from the cytosol into the intraperoxisomal membrane space.
2,18Detailed mutational analysis has been carried out in 35 unrelated ALD individuals, and all had X-q28 ALD gene mutations.
20,21 Six percent had large deletions, and 17% had an AG deletion in exon 5; the remainder had “private” mutations that were specific for each kindred, of which 55% had missense mutations and 30% had frame-shift mutations; nonsense mutations occurred in 8% and splice defects in 4%. No correlation between the nature of the mutation and the phenotype has been detected.
The ALD gene is subject to X-inactivation that has significant implications for female (XX) members of a family with an X-linked inherited disorder.
22 This process, by which one of the two X chromosomes becomes condensed and inactive, occurs at random in the embryonic stage and is permanent. If the defective allele is on the chromosome that has been inactivated, there will be no phenotypic manifestation of the disease. If, on the other hand, the defective allele is on the active X chromosome, the other having been inactivated, there will be clinical expression of the disorder.
23PATHOGENESIS OF ALD AND ITS VARIANTS
Although VLCFA accumulate in all cells, those in the adrenal cortex, the Leydig cells in the testes, and myelin-producing cells (both peripheral and central) are preferentially affected. In these tissues, the levels of VLCFA may be up to 100-fold higher in people with ALD when compared with control subjects.
24,25 There is now convincing evidence that defects in the ALD protein are the cause of ALD. Mutations in the ALD gene have been demonstrated in all ALD patients studied in sufficient detail.
2 Furthermore, transfection of ALD cells with normal chromosomal DNA has been shown to correct the defect in peroxisomal VLCFA beta-oxidation.
26Adrenal and Testicular Dysfunction: Noninflammatory Cell Death
It is believed that the ALD protein plays a role in VLCFA CoA synthetase transport into the peroxisomal membrane; thus, defects in this gene impair VLCFA degradation.
27 Transport of VLCFA into the peroxisome follows a concentration gradient. Initially VLCFA will accumulate within the peroxisome; eventually, there is an increase of VLCFA within the cytosol as well. There is some evidence that the problem of accumulation is further exacerbated by increased production of VLCFA in ALD.
28,29 These excess cytosolic VLCFA are then incorporated into lipids that accumulate within the cell as lamellae or are incorporated into the cell membrane phospholipid bilayer, which usually contains only fatty acids with 16–18 carbon lengths.
30 The incorporation of VLCFA into the cell membrane results in a structurally unstable membrane and abnormal membrane function.
In vitro it has been demonstrated that incorporation of VLCFA into the cell membrane of adrenal cortex cells results in a lack of response of these cells to adrenocorticotropic hormone (ACTH).
30,31 This nonresponsiveness is thought to occur because of a structural alteration in the cell membrane that affects expression of the ACTH receptor. Such abnormalities may also occur with other important cell membrane functions within the targeted tissues, although there is no direct evidence for this to date. The steps that lead from cellular dysfunction to atrophy and cell death have not been clarified.
32Neurological Dysfunction: Noninflammatory and Inflammatory Cell Death
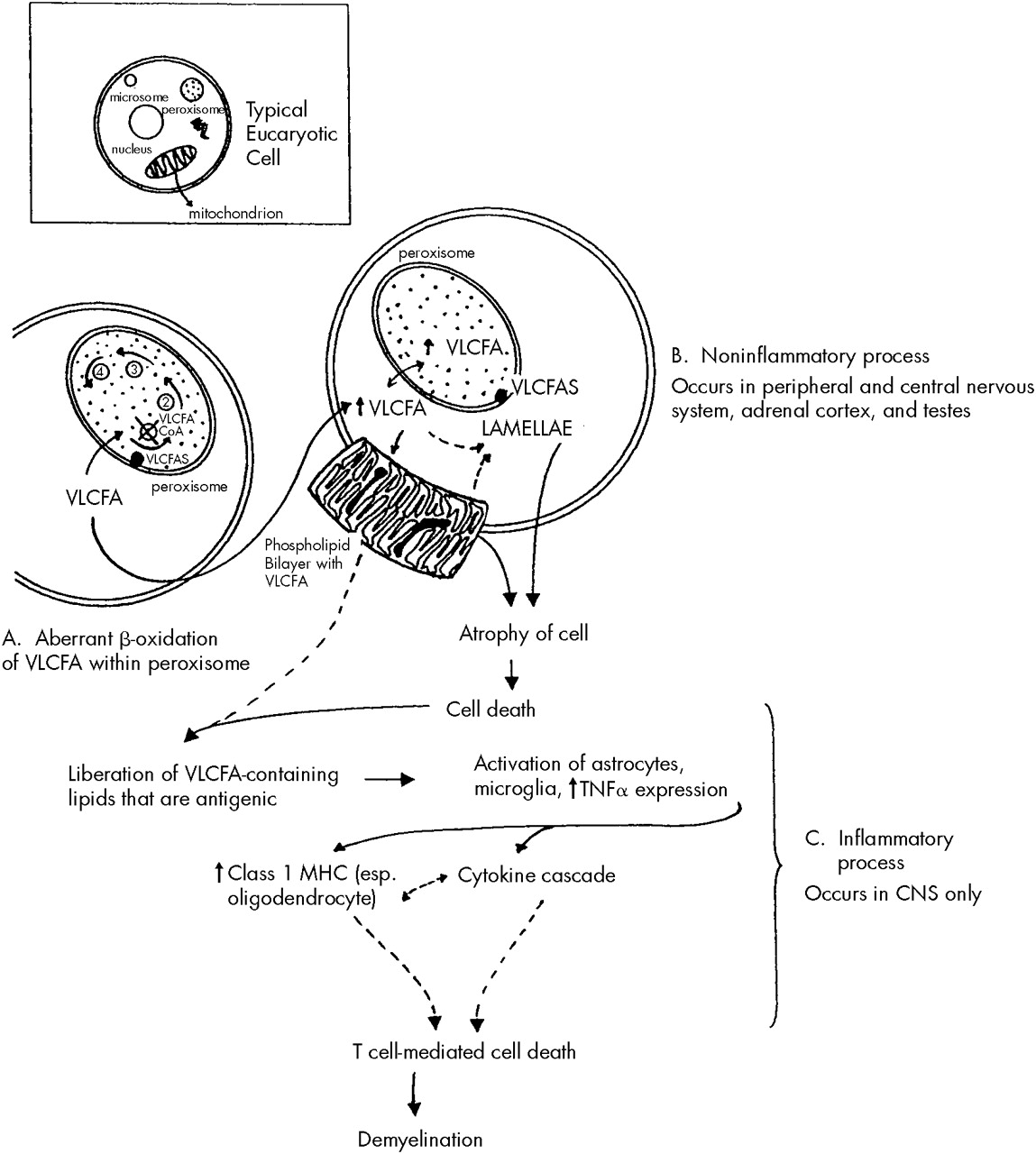
A similar process to that described above affects those cells that make myelin, namely the Schwann cells in the peripheral nervous system and the oligodendroglial cells in the central nervous system. A two-stage process may occur in cerebral ALD in the central nervous system, but not the peripheral nervous system. It begins with an initial noninflammatory dysmyelination similar to that described for the adrenal and testicular dysfunction. This process is depicted in
Figure 1. The initial dysmyelination is associated with a mild macrophage response. This can progress at some point, for unknown reasons, to a secondary phase of confluent inflammatory demyelination.
33It has been postulated that the dysmyelination occurring secondary to VLCFA incorporation into cell membranes liberates abnormal lipids that may act as triggers in ALD to initiate the cascade of inflammatory demyelination that appears to be both cytokine and T cell mediated.
33,34 Although there is a mild to moderate excess of VLCFA in all brain lipid species in ALD, the greatest increase occurs in gangliosides, phosphatidylcholine, and proteolipid protein. All of these have been shown to be antigenic and capable of activating astrocytes and microglia. Once activated, these cells produce and release tumor necrosis factor-alpha and can initiate a cytokine cascade that leads to increased expression of major histocompatibility complex class I molecules, especially on oligodendroglial cells. The presence of major histocompatibility complex class I molecules signals T cells (especially CD8 T cells) to destroy the cells on which these molecules are expressed. This process raises the possibility of oligodendroglial cell destruction by CD8 T cells, which would lead to extensive demyelination such as that seen in inflammatory cerebral ALD brains. These steps are outlined in
Figure 1. Both noninflammatory and inflammatory types of demyelination may occur within the central nervous system simultaneously. Once an immune response has been triggered, there is often a much more devastating clinical course.
The inflammatory lesions resemble those seen in multiple sclerosis. They are symmetrical, confluent white matter lesions that typically begin in the parieto-occipital regions and progress asymmetrically toward the frontal and temporal lobes. Arcuate white matter fibers, which connect intrahemispheric areas, are usually spared. The loss of myelin exceeds that of axons, but there is also considerable axonal loss. Inflammatory lesions are not commonly seen in the spinal cord other than in the corticospinal tract.
CASES IN THE LITERATURE
A review of the literature in English was conducted by using online search methods and review of citations in relevant papers. The search revealed 34 cases of adult-onset ALD that included details of the clinical examination.
9–16,34–51 We recently reported an additional case, which is included in this analysis.
51aAlthough the focus of this article is on male X-linked ALD patients, as many as 20% to 50% of female heterozygotes have neurological signs and symptoms that resemble, and may be diagnosed as, multiple sclerosis.
52 These patients usually have an onset of illness in the fourth decade, milder symptoms, and a more protracted course. Only 3% have cognitive decline and less than 1% show evidence of adrenal dysfunction.
2,53,54Age at Symptom Onset
It is often difficult to determine the precise age at which the clinical expression of a degenerative, chronic illness begins. In adult-onset ALD cases in the literature, patients are sometimes described as having been hyperpigmented since birth or early childhood, or as having “always been clumsy,” without these observations being recognized at the time as signs of a disease process. Similarly, evidence of subtle personality change or emotional lability may have been overlooked or attributed to other causes. Nevertheless, approximate ages at onset for adrenal manifestations (mean age, 29.1±6 years) and central nervous system manifestations (neurological, 31±5 years; psychiatric, 32±3 years) were provided for the majority of cases. There is great variability in the reported ages of symptom appearance, ranging from 2 to 61 years of age, reflecting the problem with the designation “adult onset.” There is no correspondence in the age at symptom onset within families; indeed, within a given kindred there are commonly childhood and adult-onset forms of the disease.
Psychiatric Features
The literature offers very little information on the incidence or prevalence of psychiatric symptomatology in patients with ALD, although one study suggests that it may be quite high.
8 Kitchen et al.
8 reviewed 109 cases of ALD, of which 6 presented after the age of 20. Of the latter, 4 had psychiatric symptoms at the time of presentation, and 1 of the 4 had only psychiatric symptoms. Unfortunately, this and most other published reports provide few specific details regarding the mental status of these patients.
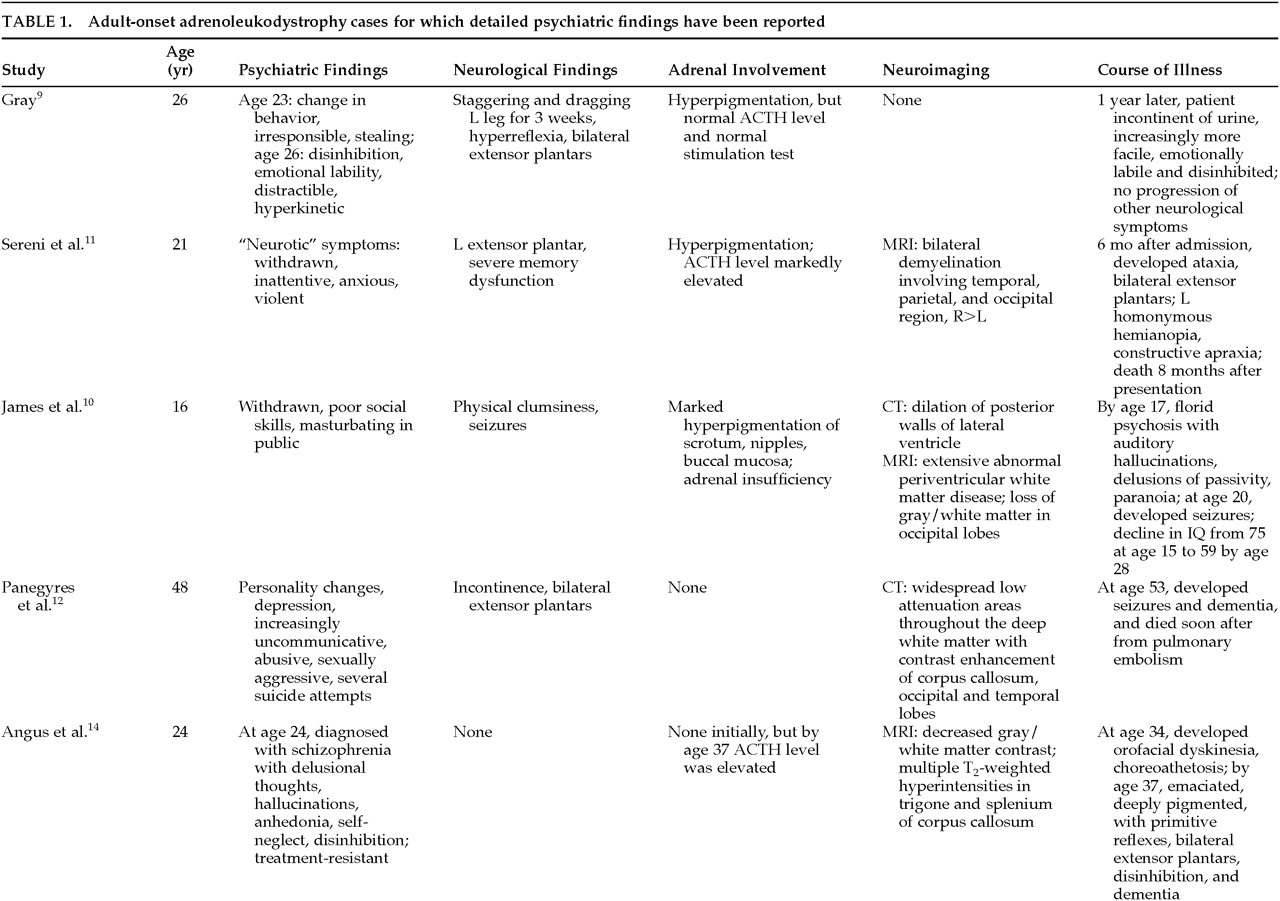
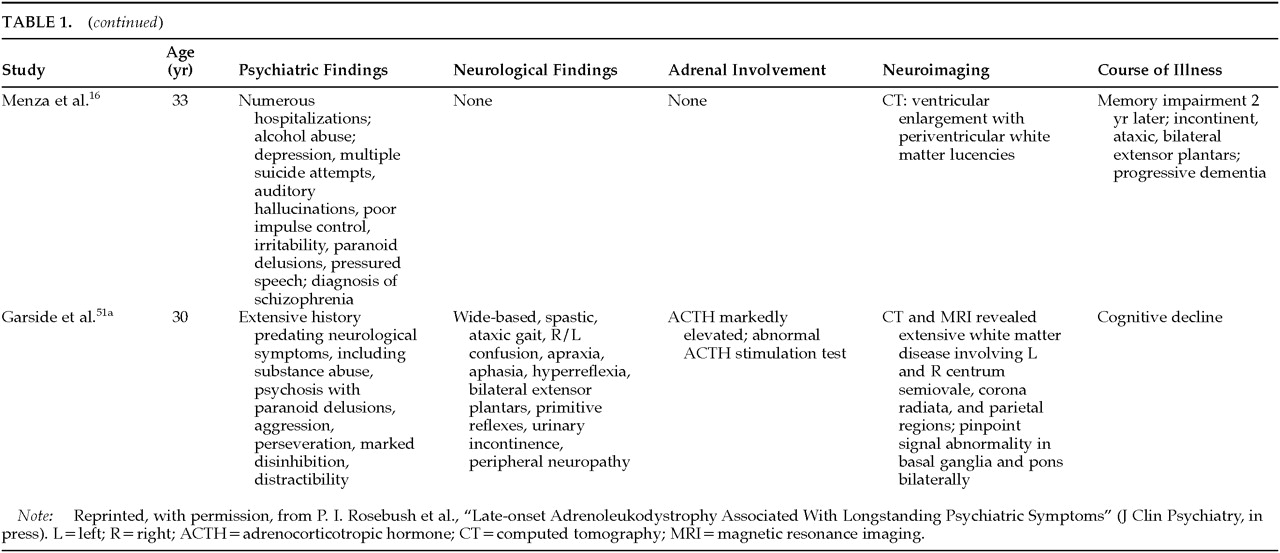
Of the 34 cases in the literature, 56% (19) were reported to have psychiatric symptomatology. In 13, a detailed psychiatric assessment is provided; the findings are summarized in
Table 1A,
Table 1B, and
Table 1C. The earliest reported symptom in 8 of these 13 patients for whom there is detailed psychiatric information was a change in behavior or personality, noted to occur anywhere from 2 months to 8 years prior to the onset of other problems. Additionally, 6 other patients for whom there is less information also had early changes in behavior. Twelve of 13 patients had symptoms of mania, including disinhibition, emotional lability, increased spending, hypersexuality, loudness, and perseveration; 9 of the 12 were given the diagnosis of bipolar disorder. In the majority of cases, the psychiatric presentation was indistinguishable from a ‘typical’ manic presentation. In addition, 5 of these patients were also psychotic. A careful review of these cases does not reveal any obvious, atypical component to their presentations. In the cases for which there is information regarding the course of illness, however, many of the patients were treatment-resistant and appeared to have an aggressive course of illness.
Many of the adult-onset ALD patients had cognitive decline. Nine of the 13 patients described in
Table 1A,
Table 1B, and
Table 1C showed clear evidence of cognitive impairment, and in most cases for which longitudinal observations are available, there was evidence of cognitive decline.
Neurological Features
Difficulty walking or a change in gait was the most common initial neurological complaint in 91% of adult-onset cases. Some patients had more than one problem that prompted a medical assessment, including seizures in 5 and peripheral neuropathy in 7 of the 34 cases.
The nature and frequency of neurological signs and symptoms are shown in
Table 2. Most commonly, patients had evidence of upper motor neuron involvement preferentially affecting the lower extremities (95%), as well as peripheral sensory involvement (58%), visual defects (21%), seizures (20%), and dysarthria (20%). In all cases there was relative sparing of upper extremities, and there was no clinical evidence of involvement of the basal ganglia or cranial nerves. A number of reports exist of ALD presenting with cerebellar involvement.
38,44,51Endocrine Abnormalities
Twenty-two of the 34 cases examined had biochemical evidence of adrenal insufficiency. The average age at the time of diagnosis of Addison's disease was 29±6 years. Of the 22 patients, 12 had clear evidence of adrenal insufficiency prior to the onset of any other symptoms, and another 8 patients were diagnosed with Addison's disease during their initial presentation with psychiatric or neurological symptoms. In 2 cases, adrenal insufficiency was diagnosed after the onset of neurological symptoms. Evidence of hyperpigmentation present since birth or early infancy was mentioned in 6 cases, including one of the cases in which adrenal insufficiency was diagnosed after the onset of other symptoms. Three patients (9%) had no clinical or biochemical evidence of adrenal involvement. Another 3 with no clinical symptoms did not receive biochemical testing, and 4 remaining patients had clinical evidence of possible adrenal insufficiency that was not investigated further. Overall, adrenal function is normal in one-third of cases at the time of clinical presentation for psychiatric or neurological problems.
2,55–57There was very little information regarding gonadal dysfunction in these cases, although in larger series, testicular dysfunction has been reported in 80% of patients.
58 Testosterone levels are usually normal, but the testosterone/luteinizing hormone ratio is significantly lower than control levels in 82%, reflecting damage to the Leydig cells. Fifty-four percent of patients with ALD will have impotence.
58Neuroimaging
The majority of patients with psychiatric or neurological findings will have evidence of demyelination on cranial MRI. Typically, T
2-weighted symmetrical hyperintensities are seen in the parieto-occipital regions. As the disease progresses, the demyelination moves forward into the frontal lobes. Some atypical findings have been described, including early frontal lobe involvement (in approximately 15% of patients), early cerebellar involvement, or an asymmetric mass lesion mistaken for a brain tumor.
59–63 In three cases the disease process appears to have been triggered by a head injury, with the pathological changes beginning in the same location as the trauma.
13,64Visual and auditory pathways were reported to be abnormal in 77% and 63% of patients, respectively. Interestingly, despite frequent MRI abnormalities in the visual pathways, visual evoked potentials were abnormal in less than 5% of ALD patients,
3 whereas brainstem auditory evoked potentials were abnormal in more than 90% of ALD patients.
4Familial Involvement
As discussed above, ALD is an X-linked recessive disorder. First-degree male relatives of affected individuals will therefore either be normal or inherit the gene defect. As noted above, 20% of women heterozygous for the ALD gene have neurological symptoms that are often diagnosed as multiple sclerosis. The course of illness in these women is much milder, and adrenal insufficiency is rare.
The same gene abnormality within a family can produce a wide range of clinical phenotypes.
46,65 For example, within one kindred studied over four generations, varying combinations of leukodystrophy, myeloneuropathy, peripheral neuropathy, and primary Addison's disease were identified in 14 of 49 family members covering an age range of 4 to 59 years and involving 7 men and 7 women. Of the 14 with these abnormalities, 7 (all male) had a childhood onset; 3 of the 7 had only primary adrenal insufficiency, and the other 4 had neurological, adrenal, and gonadal involvement. Four members of the kindred (all female) had adult-onset ALD, and of these, only 1 had neurological disease. One woman had bilateral foot drop and no other manifestations, while another had severe spastic paraparesis, sensorimotor neuropathy, and urinary urgency. Two other members, also female, had onset before the age of 1 year, with spastic paraparesis and delayed walking.
46It is striking that in all well-described adult-onset cases there is no information about the family psychiatric history.
DIAGNOSIS AND TREATMENT
Differential Diagnosis
Psychiatric illness, cognitive impairment, and neurological signs or symptoms in patients should raise the clinician's index of suspicion and lead to further investigations of an underlying metabolic disorder. It is now known that many of the metabolic diseases thought to occur only in childhood can present in milder forms in adulthood, and in these later-onset cases, psychiatric symptomatology is often prominent. Because many of these diseases are associated with an abnormal cranial MRI, this should be an initial investigation. Multiple T
2-weighted hyperintensities in the white matter, a common finding in ALD, are also seen in vitamin B
12 deficiency,
66 metachromatic leukodystrophy,
67 multiple sclerosis,
68 Krabbe's disease,
69 and solvent abuse.
70 All of these disorders can be associated with psychiatric illness and should be considered in the differential diagnosis.
71–79There are no pathognomonic physical findings associated with X-linked ALD. In addition, the neurological signs and symptoms outlined above and in
Table 2 may or may not be present in the early stages of the disease. Routine laboratory investigations are usually normal except in those patients with untreated adrenal insufficiency, who may have electrolyte abnormalities secondary to hypoaldosteronism. Patients with ALD will often have abnormal auditory evoked potentials and normal visual evoked potentials.
3,79,80 This pattern is useful diagnostically when trying to differentiate between multiple sclerosis and ALD. The finding of abnormal visual evoked potentials with normal auditory evoked potentials is more suggestive of MS. If a patient has adrenal insufficiency, impotence, psychiatric symptomatology, and evidence of upper motor neuron findings in the lower extremities, the yield from investigating for ALD would be very high.
The definitive diagnostic test for ALD is the measurement of VLCFA in serum. Most laboratories measure the absolute concentration of C26:0 as well as the C24:0/C22:0 and C26:0/C22:0 ratios.
2 Other tests that should be ordered include serum ACTH and baseline cortisol levels (24-hour urine cortisol and
a.m. and
p.m. serum levels). If either is abnormal, an ACTH stimulation test should be performed to evaluate adrenal reserve. The adrenocortical insufficiency in ALD can be life-threatening if not treated, and all patients with ALD should have a regular reassessment of adrenocortical function if the initial results are normal.
Treatment of Underlying Disorder
Following the discovery of elevated levels of VLCFA in blood and other tissues of patients with ALD, the treatment of the disorder initially focused on dietary manipulation. However, diets low in VLCFA such as C26:0 failed to lower plasma concentrations or alter the progression of illness.
81 Because there also appears to be an upregulation in VLCFA synthesis, alternative dietary treatments including Lorenzo's oil (a 4:1 ratio of oleic acid and erucic acid) were developed. These monounsaturated fatty acids competitively inhibit the fatty acid elongation system, thus decreasing production of VLCFA. Although Lorenzo's oil has been shown to decrease plasma levels of VLCFA, it has not been shown to halt the neurological progression in the cerebral forms of ALD. There is some evidence that presymptomatic patients may benefit from extended treatment.
82–84 Newer treatments under investigation include gene therapy and immunosuppression with cyclophosphamide, beta-interferon, and thalidomide.
2Bone marrow transplantation is now being used for ALD patients who have not yet demonstrated clinical symptoms but who have MRI evidence of demyelination.
85,86 The rationale for bone marrow transplantation is to provide the patient with healthy cells that have the ability to degrade VLCFA.
5 Bone marrow transplantation has been shown to reverse or stabilize abnormalities on MRI.
87 Such an approach seems to be less effective when the individual has already developed neurological findings; this makes early diagnosis and the testing of other family members crucial.
Treatment of Psychiatric Symptoms
The pharmacological treatment of psychiatric symptoms in patients with metabolic disorders is problematic. Not only do patients often not respond to medications in a typical manner, but they may be extremely vulnerable to developing side effects.
88,89 In addition, there is reason to believe that treatment with neuroleptics might actually worsen the underlying pathology in some of these disorders. Neuroleptics have been shown to inhibit oxidative phosphorylation and increase levels of free radicals in the brain.
90,91 This could, in the face of an already compromised system, decrease the ability of the brain to make adenosine triphosphate and thus further compromise affected areas, rendering them more susceptible to the disease process. Extrapyramidal side effects (EPS), particularly parkinsonism, may worsen functional impairment by adding rigidity and bradykinesia to spasticity. Akathisia can be especially troublesome to patients whose ability to move about is compromised because of neurological problems. In addition, EPS may obscure clinical recognition of changes or deterioration secondary to the underlying illness. Anticholinergic agents used to treat EPS may exacerbate cognitive impairment, dysphagia (by causing dryness), and hypotension that may be present in patients with ALD.
Atypical antipsychotic drugs such as risperidone, olanzapine, and clozapine are all reported to induce fewer EPS than typical agents such as haloperidol. They are therefore often the antipsychotic drugs of choice in the treatment of patients with neurological impairment. With respect to risperidone, however, emerging evidence suggests that it may offer little advantage over older medications in the treatment of primary psychiatric illnesses
92 or for patients with neurological disorders.
93,94 Olanzapine
95 and clozapine
96 may be effective for the treatment of psychotic or manic symptoms in ALD patients, but their anticholinergic properties will have implications for individuals who are cognitively impaired or prone to hypotension and dizziness due to adrenal insufficiency. Clozapine may be additionally problematic, given that 20% of patients with ALD develop seizures and clozapine is known to precipitate seizures in 3% to 5% of patients. One study reported a lifetime cumulative risk of seizures as 10% after 3.8 years of treatment.
97 This is not in itself a contraindication to its use, given that the risk of seizures is dose-related and they can be prevented or effectively treated with either lower dosages or the concurrent use of antiepileptic agents. If clozapine were prescribed, it would be prudent to obtain an EEG prior to treatment and following achievement of the optimal dosage. Olanzapine is not known to lower the seizure threshold.
95Given the frequency of manic symptomatology in psychiatrically disturbed patients with ALD, mood stabilizers may be effective. There is no information available about whether one medication is more effective or has a better side effect profile than another in these patients. Clinicians prescribing lithium will have to keep in mind the potential for ALD to be associated with electrolyte disturbances secondary to hypoaldosteronism, which may affect drug reabsorption and, therefore, serum levels. Benzodiazepines may control manic symptoms or behavioral disturbances, although they can worsen ataxia. Their central muscle-relaxant properties may be of benefit in relieving spasticity.
The essential underpinnings of all appropriate pharmacological treatment decisions are a thorough knowledge of the drugs being prescribed and an understanding of the primary disorder, particularly the ways in which it might render the patient vulnerable to certain side effects.
SUMMARY
Although this review has focused on the neuropsychiatric aspects of ALD, the most important point to be made is that all psychiatric patients need to be assessed thoroughly at the time of admission and again over time. Assessment should always include a detailed family history of medical and neurological disorders, a neurological examination, and neuroimaging. When a patient develops neurological findings, becomes treatment-refractory, or has cognitive decline, consideration should be given to unusual causes of psychiatric illness, such as metabolic diseases. These diseases are typically considered to be of childhood onset and are therefore most certainly underrecognized in adulthood. Awareness and recognition of diseases such as ALD have important implications for family genetic counseling and treatment considerations. In addition, because all psychiatric diagnoses are syndromes defined by symptom complexes, it is important to identify those subgroups of patients whose psychiatric symptoms are better explained by other diseases, so that the genetics and natural history of psychiatric disorders can be better understood.