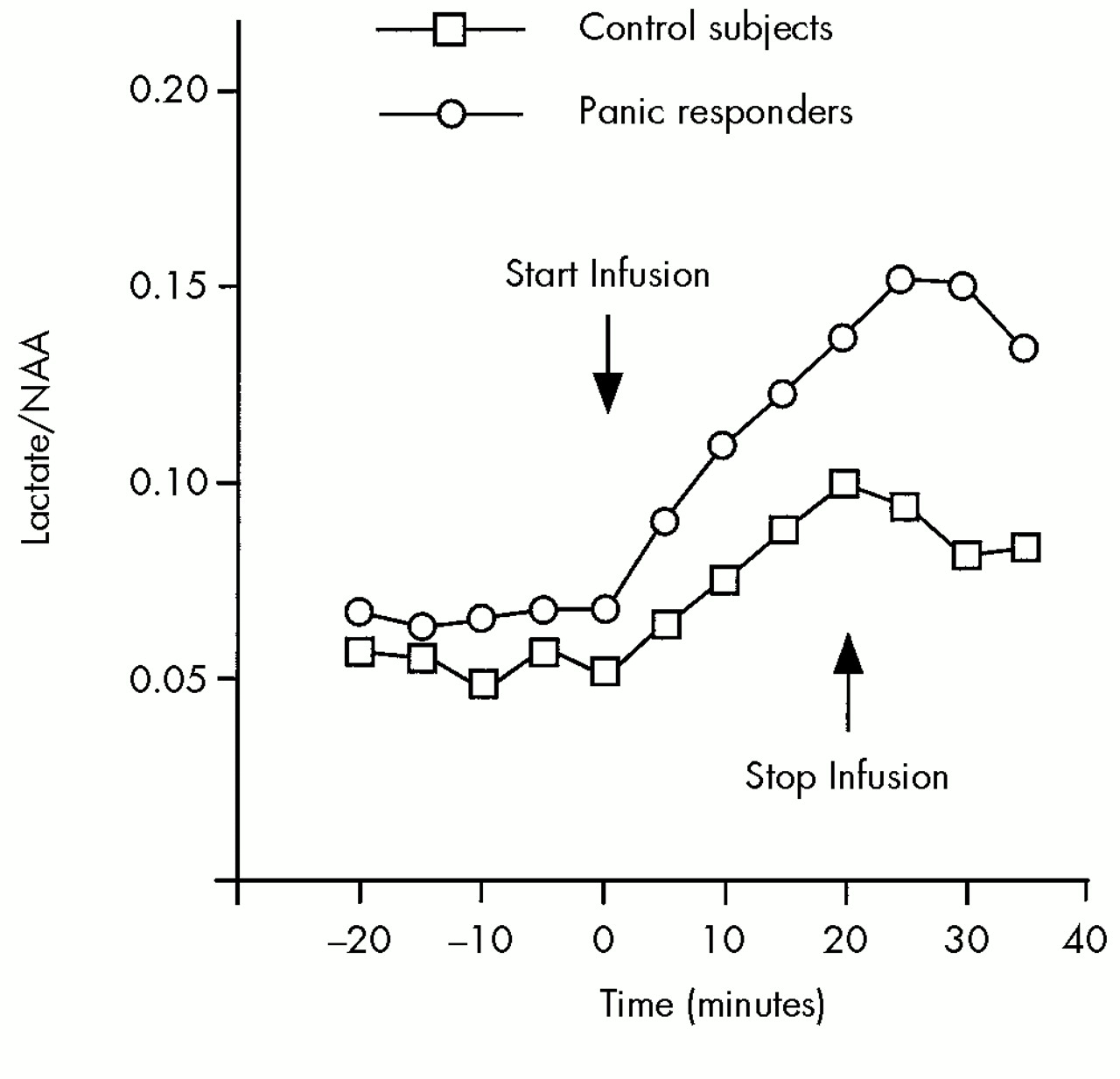
A variety of physiological or pathophysiological mechanisms may contribute to the exaggerated lactate response to alkalosis observed in panic disorder. Identifying the specific mechanism of this abnormal response may increase our understanding of the underlying pathophysiology of panic disorder. Three possible mechanisms suggested by investigators demonstrating this effect are 1) hypoxia secondary to cerebral vasoconstriction;
10,12,13,35 2) a disturbance in the regulation of intracellular pH;
8,10 and 3) increased adrenergic activity.
8 The mechanisms are summarized in
Table 1. The following discussion examines the possible contributions of each of these mechanisms to the exaggerated lactate response to alkalosis observed in patients with panic disorder.
Cerebral Vasoconstriction and Brain Hypoxia
Cerebral blood flow changes rapidly in direct relationship to changes in arterial pCO
2. One function served by this response is to counteract changes in brain pH, such as those occurring during hyperventilation-induced alkalosis.
22 Hyperventilation reduces cerebral blood flow (CBF) by 40% to 50%.
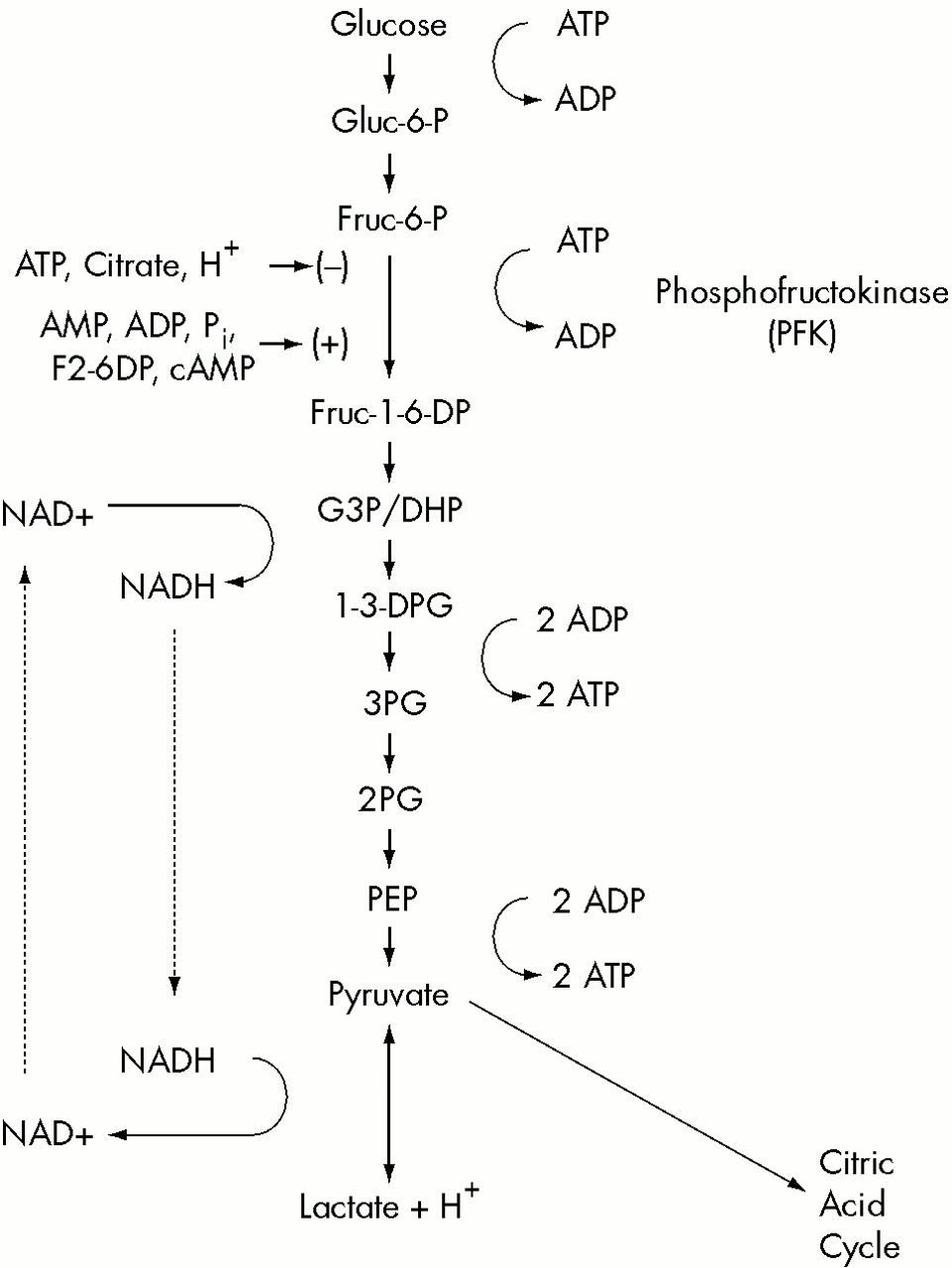
36 However, in both humans and animals, this decrease in CBF does not usually cause metabolic changes indicating brain hypoxia. The increase in brain lactate caused by hyperventilation appears to result from the direct effects of alkalosis on glycolytic flux.
19,23,36–38 However, it is possible that panic disorder patients have an abnormal vasoconstrictor response to alkalosis that leads to brain hypoxia.
During hyperventilation, CBF reaches its minimum at an arterial pCO
2 of approximately 25 to 30 mm Hg.
36 Although lower arterial pCO
2 levels may not further reduce CBF, they impede oxygen delivery by the Bohr effect (the shift in the oxyhemoglobin dissociation curve to the left with increasing pH). Clinically, hyperventilation can lead to diminished mental function, slowing of the EEG, paresthesias, muscle contractions, and, in the extreme case, loss of consciousness. These effects are often interpreted as signs of progressive brain hypoxia. However, as summarized by Edvinsson et al.,
36 “the clinical and experimental evidence for brain hypoxia is somewhat scant.” Reviews of experimental studies of brain metabolism during hyperventilation have concluded that hyperventilation leading to a pCO
2 as low as 14 mm Hg does not produce brain hypoxia.
19,37,38 The subjective effects of hyperventilation may be the result of altered function of the numerous receptors, channels, transporters, and enzymes in neurons that are highly sensitive to increases in pH, rather than the result of hypoxia.
39In attempting to determine the cause of increased lactate production in response to respiratory alkalosis, Siesjo
19 noted that it is difficult to distinguish between lactate production that occurs as a result of increased intracellular pH activating PFK and lactate production that occurs as a result of decreased oxygen delivery and cellular hypoxia. He concluded that “the most direct way of testing whether or not hypoxia is present is to measure concentrations of labile phosphates in the tissue.” van Rijen et al.
40 measured brain lactate with proton MRS and additionally measured ATP, ADP, AMP, P
i, PCr, and pH with phosphorus-31 MRS in human volunteers who hyperventilated to an average pCO
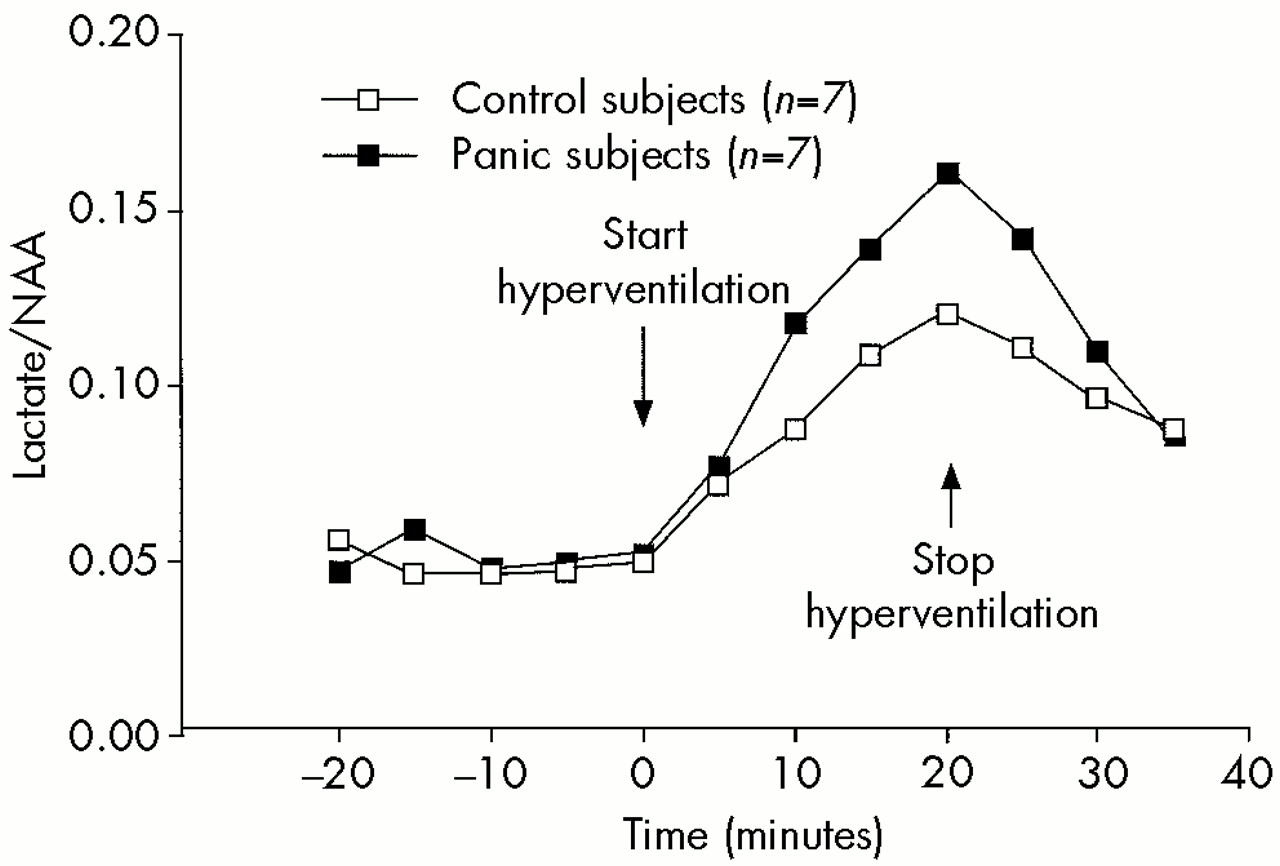
2 of 16 mm Hg. Concurrent transcranial Doppler sonography showed a 42% reduction in left middle cerebral artery flow. Brain lactate increased significantly during and after hyperventilation, but no change was observed in the phosphate compounds, suggesting that hypoxia did not occur.
Hyperventilation causes a large increase in arterial pH but only a small increase in brain pH.
40,41 The relative stability of brain pH results from homeostatic processes, which serve to counteract changes in intracellular pH. One of the most important mechanisms of this defense against intracellular alkalosis in the brain is the increased production of lactic acid via glycolysis.
19,37 This mechanism can operate in the presence of fully adequate oxygen tension. Numerous biochemical studies
42–47 and more recent spectroscopic studies
40,41 have shown that high-energy phosphate levels (ATP and PCr) remain unchanged during hyperventilation that produces pCO
2 levels of approximately 14 mm Hg. Only one study, which involved severe hypocapnia in rats, (pCO
2=10 mm Hg) found evidence for decreased PCr and increased ADP, indicating that metabolically significant hypoxia had occurred.
48 Studies of cerebral oxygen consumption (CMRO
2) during hyperventilation also typically show no reduction in CMRO
2.
47,49,50 Overall, the evidence suggests that respiratory alkalosis leading to a pCO
2 of ≥14 mm Hg produces a rise in brain lactate as a result of increased glycolysis in the service of regulating intracellular pH, and that brain hypoxia does not occur.
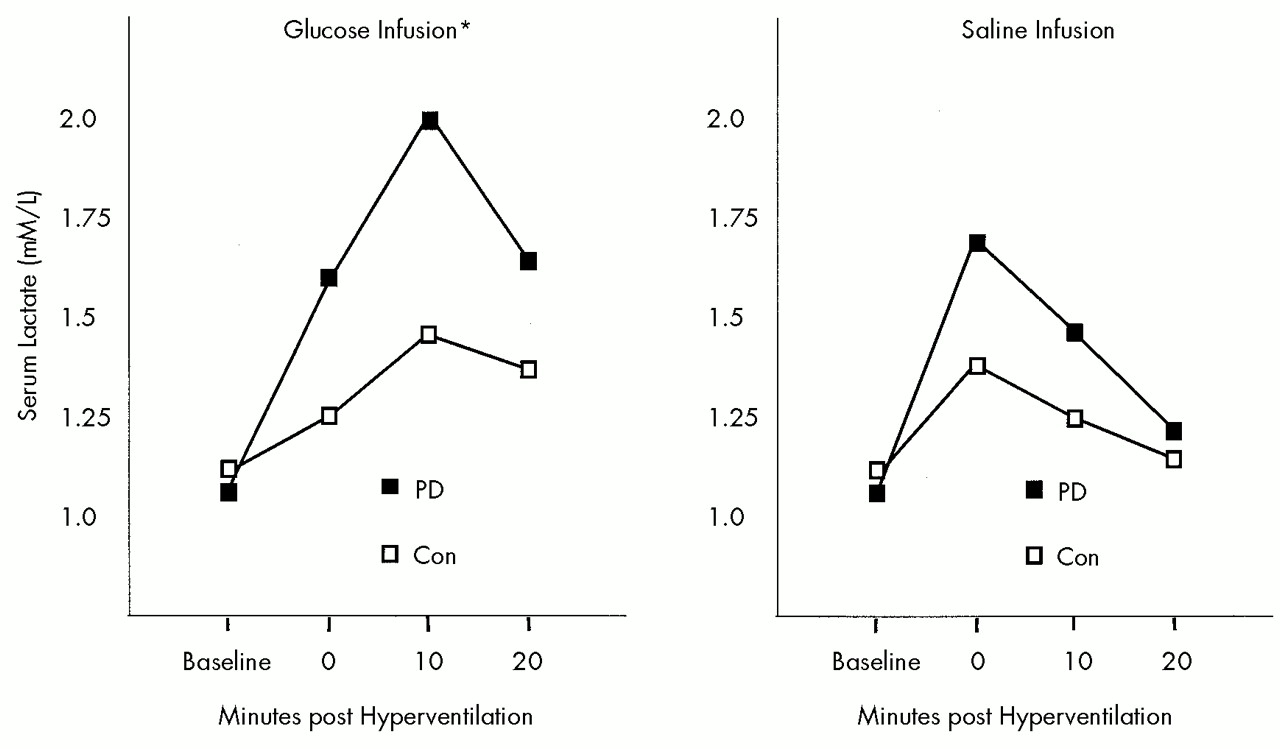
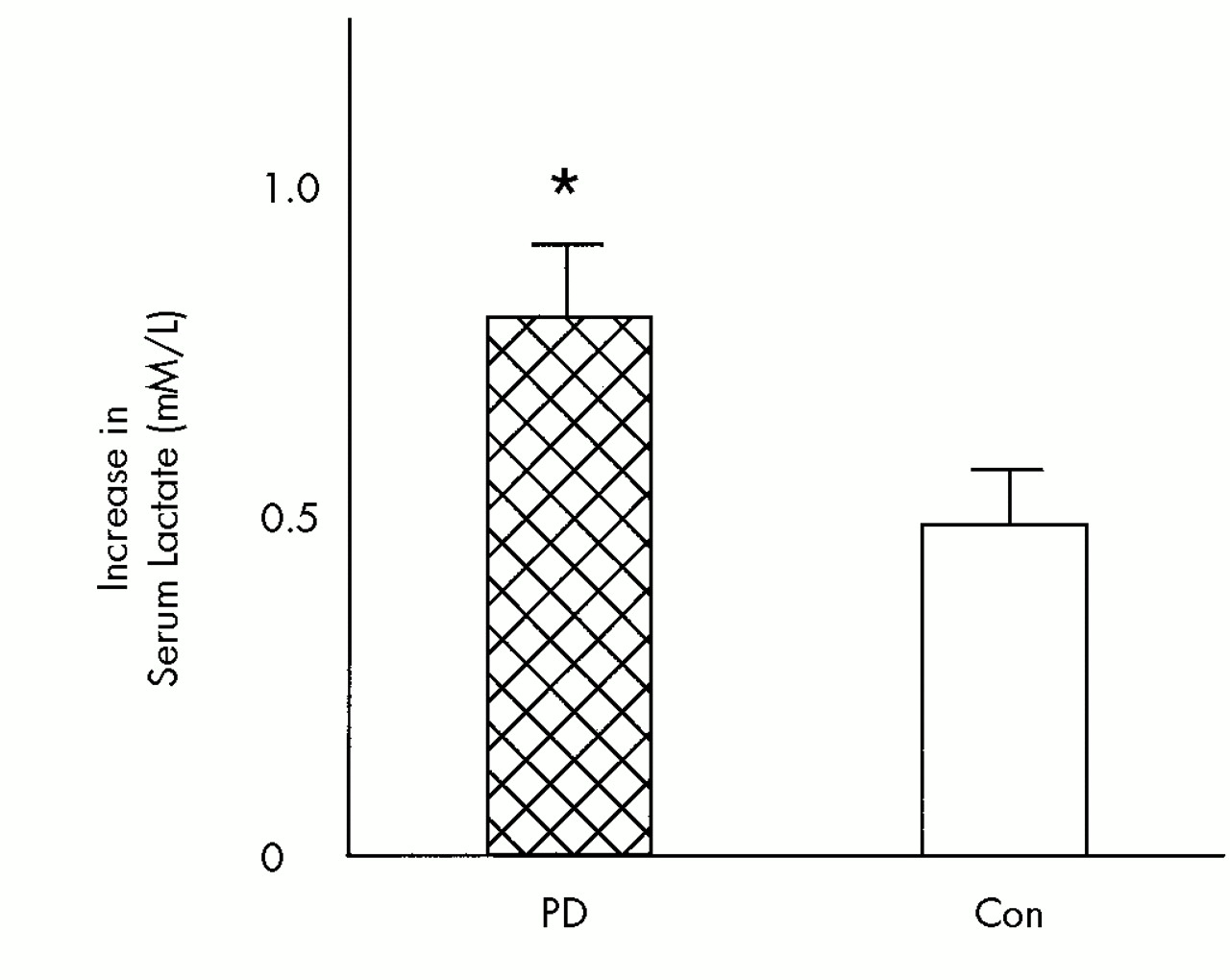
However, it is possible that an abnormal cerebrovascular response to alkalosis in panic disorder could render such patients distinctly vulnerable to brain hypoxia during hyperventilation. Recent studies have produced preliminary evidence for an abnormal vasoconstrictor response to respiratory alkalosis in patients with panic disorder. Two studies using transcranial Doppler ultrasonography of basilar artery blood flow found that panic patients have a greater than normal reduction in basilar artery flow during moderate hyperventilation (pCO
2=25 mm Hg).
51,52 However, there is not yet any evidence that metabolically significant hypoxia occurs in panic patients during hyperventilation. Nonetheless, a vulnerability to cerebral hypoxia during mild to moderate respiratory alkalosis could contribute to many of the characteristic manifestations of panic disorder.
Phosphorus-31 MRS studies of high-energy phosphates could provide an experimental test of the hypothesis that respiratory alkalosis leads to metabolically significant brain hypoxia in panic patients and not in normal subjects. Using this technique in normal volunteers, Van Rijen et al.
40 have shown that ATP, ADP, AMP, P
i, and PCr do not change with hyperventilation (to a pCO
2 of 15.8 mm Hg). If ATP and PCr decreased while ADP, AMP and P
i increased in response to a similar degree of hyperventilation in panic disorder patients but not in normal subjects, this would demonstrate an abnormal vulnerability to brain hypoxia in response to respiratory alkalosis in panic disorder.
A Disturbance in the Regulation of Intracellular pH
Intracellular pH is one of the most tightly regulated parameters in the mammalian nervous system. The maximum change in H
+ concentration that can be tolerated is approximately 0.00005 mM.
22 H
+ concentration changes beyond this range can disrupt the tertiary structure, and thus the function, of cellular proteins. Intracellular pH is controlled by a variety of regulatory mechanisms, including the increased production of lactic acid in response to alkalosis. The exaggerated lactic acid response to alkalosis in panic disorder could result from a metabolic disturbance that increases the activity of this pH regulating mechanism. For many illnesses, including panic disorder, experimentally demonstrated physiological disturbances often represent compensatory and adaptive responses to underlying, and perhaps less easily observed, abnormalities in function.
53 From this perspective, the increased lactic acid production could represent a compensatory response to a deficiency in one of the other homeostatic mechanisms protecting intracellular pH.
A variety of different mechanisms could cause a compensatory increase in lactic acid production during alkalosis. Intracellular pH is regulated by a combination of three mechanisms: 1) physicochemical buffering, 2) transport of H
+ (acid) or HCO
3– (base) across the cell membrane, and 3) metabolic processes generating or consuming H
+ ions. Increased glycolytic production of lactic acid is the most important example of the last mechanism.
22,39,54 A relative deficiency in one of the buffering or transport mechanisms protecting intracellular pH could lead to a wide range of abnormalities in the response of panic patients to disturbances of intracellular pH. These could include altered responses to acidotic as well as alkalotic challenges, such as the increased sensitivity to hypercapnia often noted in panic disorder.
1,35 The presence of a pH regulating deficiency of this type in panic disorder could, in principle, be detected by a phosphorus-31 MRS study of the intracellular pH response to hyperventilation. In this general model, the increased lactic acid production results not from hypoxia, but from a deficiency in one of the mechanisms protecting against alkalosis. The model predicts that during hyperventilation, panic patients compared with normal subjects would not show a decrease in high-energy phosphates and would have a slower or less complete correction of hyperventilation-induced intracellular alkalosis. This effect might be most apparent under conditions that limit lactic acid production, such as substrate depletion by prolonged hyperventilation in fasting subjects. This depletion would limit the extent to which increased lactic acid production could compensate for the hypothesized deficiency in intracellular pH regulation.
This model posits a deficiency in one of the buffering or transport mechanisms that regulate intracellular pH. The most important intracellular physicochemical buffer is the CO
2/HCO
3– system. Additional intracellular buffering capacity results from the imidazole groups on histidine residues of proteins and the free and bound forms of phosphate (including inorganic phosphate, AMP, ADP, and ATP).
55 A deficiency in one of these buffers could lead to a greater reliance on lactic acid production in the homeostatic response to intracellular alkalosis. At present, however, there is little evidence of deficiencies in these intracellular buffers in panic patients. In a phosphorus-31 MRS study of frontal cortex in treated patients with panic disorder and control subjects under “resting” conditions (without any controlled psychological or physiological manipulations), Shioiri et al.
56 found no significant differences in levels of any phosphate compounds.
Multiple active and passive mechanisms for transporting H
+ or HCO
3– across the cell membrane have been identified in brain parenchyma. These mechanisms modify intracellular and interstitial pH in a reciprocal manner. In neurons and glia, passive entry of H
+ through the glutamate-gated cation channel and passive efflux of HCO
3– through the GABA-gated chloride channel may slightly acidify the neuron and alkalinize the interstitial fluid.
57 Multiple active transport mechanisms that help regulate intracellular pH have been demonstrated in neurons and glia.
58,59 Four types of Na
+/H
+ exchanger (NHE) have been found in the brain.
58 The NHEs alkalinize the neuron by exchanging intracellular H
+ for extracellular Na
+. The extrusion of H
+ by NHEs is activated by agonist binding to both beta and alpha adrenoreceptors.
60–62 Another pH regulating transport mechanism, the Cl
–/HCO
3– exchanger, is particularly important in the recovery of cortical neurons from intracellular alkalosis.
63 This anion exchanger also helps regulate the smooth muscle vasoconstrictor response to alkalosis
64 and the respiratory response to hypercapnia.
65 Intracellular pH is also regulated by a H
+/ lactate cotransporter, which influences pH in both neurons and glia,
58,59 and by an Na
+/HCO
3– cotransporter, which is specific to glia. The latter mechanism may have an important role in linking glutamate uptake to glial alkalinization and increased production of lactic acid. All of these active transport mechanisms can operate in both directions, to either acidify or alkalinize neurons.
58 Dysregulation of one of the pH regulating transport mechanisms in patients with panic disorder could lead to an exaggerated, compensatory lactate response to alkalosis, as well as other abnormal responses to changes in pCO
2 and intracellular pH.
It is important to note that an exaggerated cerebral vasoconstrictor response to hyperventilation could itself result from a disturbance in pH regulation. Intracellular and perivascular pH of the smooth muscle cells lining the cerebral arterioles are principal determinants of the cerebral vasoconstrictor response to hyperventilation.
36 An exaggeration of this vasoconstrictor response, which preliminary evidence suggests occurs in panic disorder, could result from a dysregulation of one of the homeostatic mechanisms, such as the Cl
–/HCO
3– anion exchanger, which normally limit the hyperventilation-induced increase in pH within arteriolar smooth muscle cells.