The apolipoprotein E epsilon 4 allele (APOE E4) is a well-known risk factor for late-onset familial Alzheimer's disease (AD)
1 as well as for sporadic AD,
2–4 and the inheritance of the APOE E4 allele is believed to lower the age at onset in a dose-dependent manner.
5 Previous studies have strongly suggested that APOE E4 might play an important role in the pathogenesis of AD. APOE E4 may be associated with a faster deposition of the amyloid β protein,
6 and isoforms-specific interactions of APOE with the tau protein
7 and cholinergic function
8 may exist. Furthermore, several lines of clinical studies have reported that more severe cognitive,
9,10 gross structural,
11,12 and electrophysiological
13,14 abnormalities were associated with the presence of APOE E4.
By use of [
18F]fluorodeoxyglucose positron emission tomography (PET), characteristic abnormalities of the cerebral metabolic rate of glucose metabolism (CMRglc) in the temporal, parietal, cingulate, and sometimes frontal regions have been observed in patients with AD
15,16 as well as in persons who are at risk for AD.
17 In studies concerning the influence of APOE E4 on CMRglc, APOE E4 carriers showed a more pronounced reduction of CMRglc in the temporal, parietal, and prefrontal regions in normal elderly subjects with a higher risk of AD.
18,19 However, later PET studies have repeatedly failed to demonstrate the association of APOE E4 allele with further reduction of CMRglc in patients with AD.
20,21Although the pathogenic role of APOE E4 in AD has been readily recognized, there is still controversy as to whether APOE E4 affects the progression of AD. Some prospective studies have reported that there was no difference in the rate of cognitive decline in relation to the inheritance of APOE E4.
22,23 These results suggest that APOE E4 might be involved in the development of AD but does not influence the course of AD once the disease has developed. Therefore, it is possible to hypothesize that the effect of APOE E4 on CMRglc in AD might vary at different stages and that the change of CMRglc associated with the inheritance of APOE E4 is prominent only in the early stage of AD, disappearing with the progression of the disease. We suspected that the subjects with AD in previous PET studies were heterogeneous in clinical severity; however, no clear description was given.
20,21In this study, therefore, we tried to observe the effect of APOE E4 on CMRglc in patients with AD after classifying the subjects into clinically defined different severity groups. The objective of this study was to demonstrate whether the effect of APOE E4 on CMRglc is present in subjects with early-stage AD and disappears in those at later stages of the disease.
RESULTS
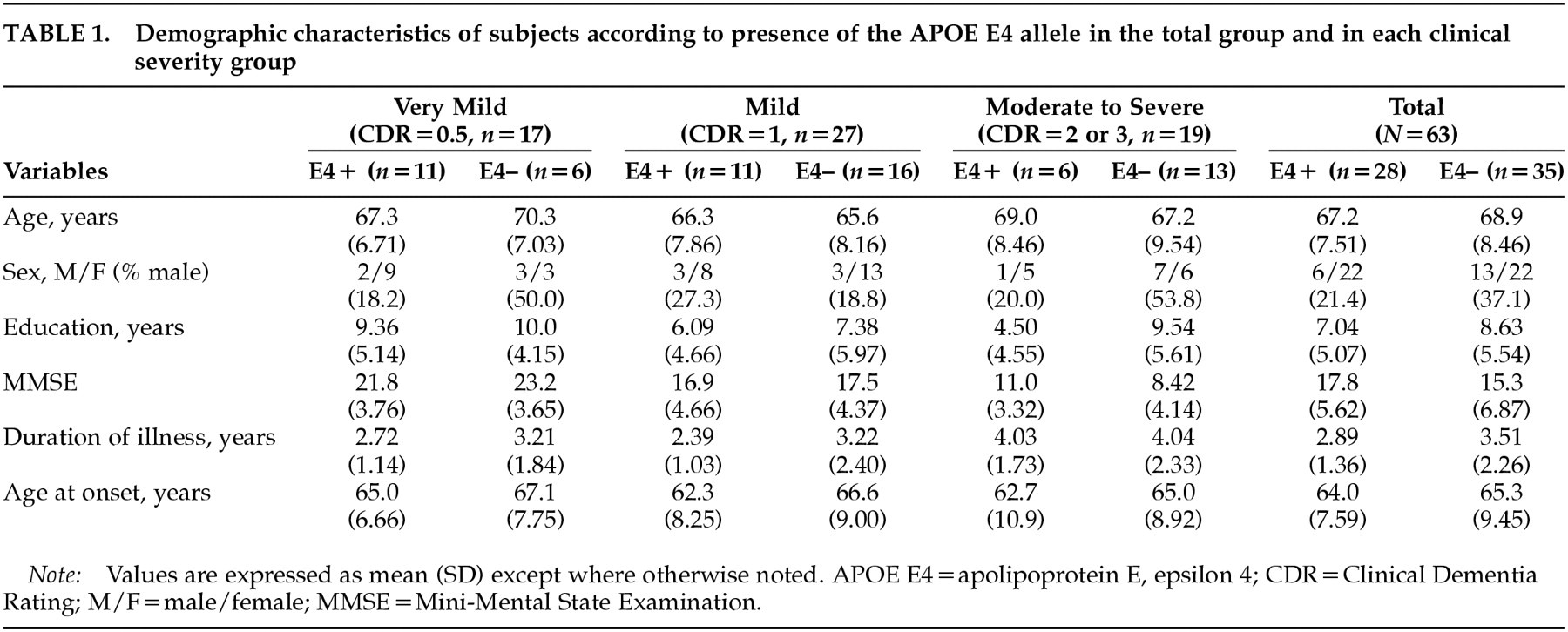
A total of 63 patients with AD participated in this study. Twenty-eight of them were APOE E4 carriers and 35 were noncarriers. There were no significant differences on two-tailed
t-tests (means for age, education, MMSE-K score, duration of illness, and age at onset) or chi-square tests (sex ratio) between the APOE E4+ and APOE e4– groups in total, or in any of the clinical severity groups (
Table 1). Although there was a tendency to lower age at onset in the APOE E4+ groups, the differences were not statistically significant.
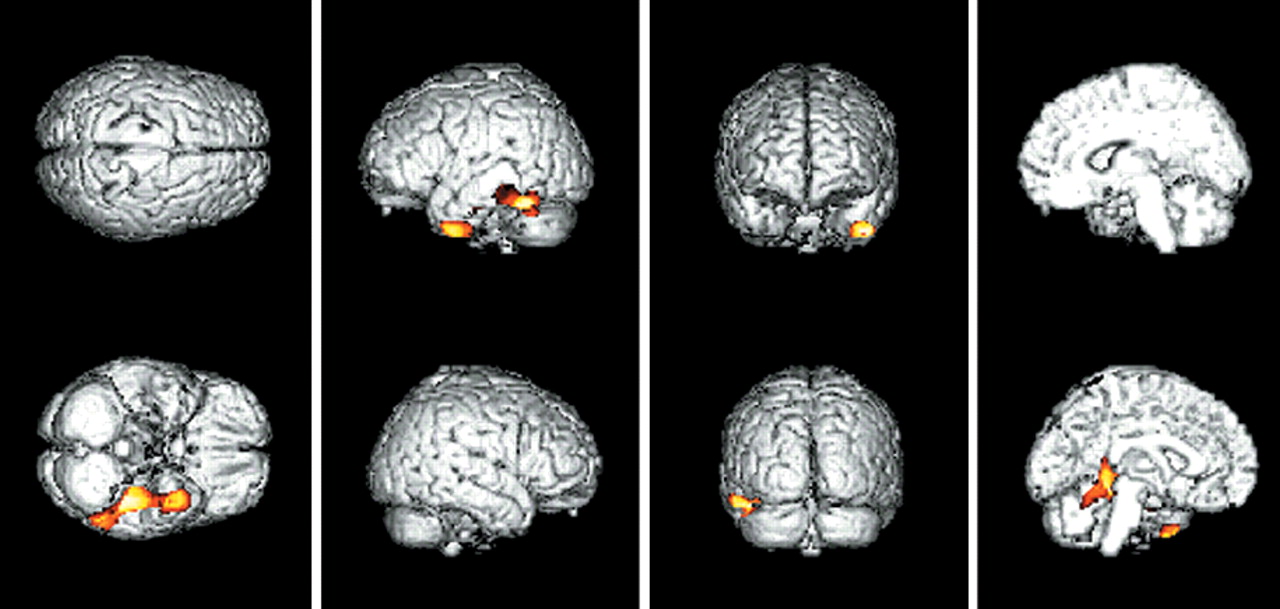
In comparing CMRglc between the APOE E4+ group and the APOE e4– group among the total number of AD patients, there was no voxel with a significant difference in glucose metabolism on SPM analysis. However, when the analyses were performed in each clinical severity group, regions where the APOE E4+ group showed greater decline of CMRglc than the APOE e4– group were observed. In the very mild clinical severity group, SPM showed a significantly decreased CMRglc in the left inferior temporal regions (3,247 voxels,
P<0.005, uncorrected for multiple comparison) in the APOE E4+ group compared with the APOE e4– group. The left inferior temporal regions included three peaks of maximal decline of CMRglc, and each peak corresponded to Brodmann areas 20, 37, 37 (
P=0.0002,
P=0.0008,
P=0.0013, respectively). The SPM results and coordinates of maximal decline are shown in
Table 2 and
Figure 1. There was no brain region in which the APOE e4– group showed greater decline of CMRglc than the APOE E4+ group in the patients with very mild clinical severity. In the mild group as well as the moderate-to-severe group, there were no regional differences in CMRglc between the APOE E4+ and APOE e4– groups.
DISCUSSION
The association of APOE E4 with greater deficits of CMRglc has been consistently reported in normal elderly subjects at high risk for AD.
18,19 However, previous studies attempting to elucidate the effect of APOE E4 on CMRglc in patients with AD have failed to do so.
20,21 Corder et al.,
21 studying the rCMRglc in 31 patients with AD, reported no association of APOE E4 allele with specific deficits in brain metabolism. The clinical severity of their study population was heterogeneous, in that 12 of the 31 subjects had only mild memory complaints. In a study of 83 AD patients by Hirono et al.,
20 the clinical severity was not discussed in great detail. These results, in fact, are consistent with our results in that no difference in CMRglc between the APOE E4+ and APOE e4– groups among the total number of AD patients was observed. In our study, 17 of the 63 subjects had very mild dementia. Therefore, the breakdown of our subjects was similar to that in the Corder et al.
21 study.
When further comparisons of CMRglc between the APOE E4+ and APOE e4– groups were conducted after classifying the subjects into different clinical severity groups, the APOE E4+ group showed greater reduction of CMRglc in the left inferior temporal region, but only in the subjects with very mild clinical severity. No difference in CMRglc was observed in the mild and moderate-to-severe groups. The subjects with very mild dementia described in our study clearly met the NINCDS-ADRDA and DSM-IV criteria for probable AD. Therefore, they differed from the subjects described in the Small et al. study,
19 who were normal elderly people at risk for AD who met the diagnostic criteria for age-associated memory impairment, but not for AD. Our results provided direct evidence that the APOE E4 allele influences the dysfunction of the brain in AD.
Although it is readily recognized that APOE E4 lowers the age at onset and increases the risk of AD, there is still controversy as to whether APOE E4 influences the rate of progression of AD. Several studies have reported that the APOE genotype did not influence the rate of cognitive decline in AD.
22,23 However, another study reported accelerated cognitive decline in APOE E4 homozygotes with AD.
32 Still other studies have reported even better prognoses in APOE E4 carriers.
33,34 The results of our study favor the hypothesis that APOE E4 is not associated with the progression of AD, but is related only to the developmental stage of the disease.
The finding of APOE e4– related hypometabolism in the left temporal region corresponds with clinical, magnetic resonance–based imaging, and neuropathologic studies. Several studies have shown that the presence of APOE E4 in AD was related to more severe impairment of the types of memory that are the function of the hippocampus and temporal lobe structures, such as delayed recall.
9,10 There have been at least two MRI studies in which a larger volume loss of the medial temporal lobe structures was seen in patients with AD homozygous for APOE E4.
11,12 In a neuropathologic study, an increased deposition of the amyloid β peptide
35 and decreased choline acetyltransferase activity
36 of the temporal lobe were reported to be associated with the presence of the APOE E4 allele.
The methodological considerations in this study include the statistical issues in analyzing the PET data by using SPM96. Although a successful clinical validation study using SPM was reported recently,
37 the danger of increased alpha error due to multiple comparisons still exists. Because a standard guideline for proper acquisition of statistical inference has not yet been established, we applied a commonly used setting (
P<0.005, SPM [
Z]>2.58) for explorative purposes. However, the SPM (
Z) score of the observed cluster (left inferior temporal region) was actually more than 3.09 (
P<0.001). Therefore, it is less likely that significant clusters were observed by chance.
In this study, we showed that the presence of the APOE E4 allele was associated with greater reduction of cerebral glucose metabolism in patients with AD. Furthermore, reduction of glucose metabolism was prominent only in the very early stage of AD; the effect of APOE E4 on CMRglc disappeared in the more advanced stages of the disease. These results confirm that the deficit of CMRglc in AD is influenced by APOE E4 and favor the hypothesis that APOE E4 is mainly related to the development of AD, but not to its progression. Further longitudinal studies are necessary to determine the effects of APOE E4 on disease progression of AD.