Metachromatic leukodystrophy (MLD) is a demyelinating genetic disorder in which neuropsychiatric abnormalities, including psychotic features similar to those present in schizophrenia, are prominent symptoms. Although rare, it is a valuable model for the study of psychosis.
2 MLD occurs worldwide with an estimated frequency of 1/40,000, although several clusters of much higher frequency have been identified.
3 MLD is caused by an autosomal recessive mutation on chromosome 22q, resulting in the near-complete absence of the lysosomal enzyme arylsulfatase A (ARS-A). More than 57 mutations of the ARS-A gene have been identified, including 31 amino acid substitutions, 1 nonsense mutation, 3 deletions, 3 splice donor site mutations, and 1 combined missense-splice donor site mutation.
3,4 There is also an ARS-A pseudodeficiency, with a frequency of around 16% in the general population.
5 These patients, who appear clinically healthy, are thought to be compound heterozygotes for the deficient allele and another allele coding for low enzyme activity. MLD can be mimicked by genetically unrelated deficiency of cerebroside sulfatase activator and multiple sulfatase deficiency.
Metachromatic material (cerebroside sulfate) accumulates in brain oligodendrocytes and microglia, peripheral nerve Schwann cells, kidneys; and it is excreted in urine. The accumulation of this material leads to abnormal myelin formation and myelin degeneration by unknown pathophysiological mechanisms. Diagnosis of MLD may be suspected when metachromatic granules are found in conjunctival or sural nerve biopsy.
6 Definitive diagnosis relies on the confirmation of low ARS-A activity in leukocytes or cultured fibroblasts.
3 The clinical phenotype of MLD is divided into late infantile, juvenile, and adult-onset forms. The late infantile form, which is the most common, presents before age 2, with weakness, muscular hypotonia and delayed motor and intellectual milestones. Death usually occurs before age 5 in a rigid, vegetative state. Juvenile MLD most commonly presents between 4 and 10 years of age. The distinction between juvenile and adult-onset MLD is arbitrarily set at age 16.
Both the juvenile and adult-onset forms are distinguished from late infantile MLD by slower progression, higher residual ARS-A activity, and a preponderance of neuropsychiatric abnormalities, including conduct disorder, nonverbal learning disability, alcohol or substance abuse, depression, and schizophrenia-like psychosis.
3,7,8 Baumann et al.
7 described two distinct clinical patterns of adult MLD.
7 One presents with predominantly motor manifestations similar to those found in infantile cases. The other presents as a schizophrenia-like psychosis, with prominent negative signs (e.g., psychomotor slowing, apathy, apragmatism, poor judgment, and disorganized thinking). The cognitive profile resembles a subcortical dementia, including impaired verbal fluency, impaired memory retrieval, and executive dysfunction. Disinhibition, anosognosia, complex delusions, bizarre behavior, and auditory hallucinations may also occur.
7,9 Extrapyramidal symptoms, including choreoathetosis and dystonia, seizures, and peripheral neuropathy rarely occur. Some variability is probably related to different mutations within the ARS-A gene. For example, adult MLD patients with primarily psychiatric manifestations may carry mutations in regions of the ARS-A gene that are distinct from the common P426L mutation found in most patients with motor signs.
7 However, strict genotype-phenotype correlations do not apply in all cases, and genetic background probably modifies the clinical profile.
4 Whatever the age of onset, progression is the rule. Motor signs can be absent until decades into the illness despite the characteristic white matter appearance on neuroimaging.
10 The prominent behavioral disturbance and more subtle initial cognitive dysfunction combined with the lack of motor signs can make recognition of this degenerative dementia challenging. All cases eventually manifest dementia and spasticity.
It is unclear at this time whether the neuropsychiatric profile of adult MLD is a direct manifestation of low ARS-A activity or whether neuropsychiatric abnormalities, particularly psychosis, reflect disruption of brain white matter tracts. In favor of the low-ARS-A-activity-neuropsychiatric-phenotype model is the observation of increased psychopathology in some first-degree relatives of MLD patients. These individuals are presumably heterozygote carriers of one defective allele of the ARS-A gene.
8 Subtle deficits in visuospatial perception, construction, stereognosis, and visual search tasks have been found in MLD heterozygotes.
11STRUCTURAL IMAGING
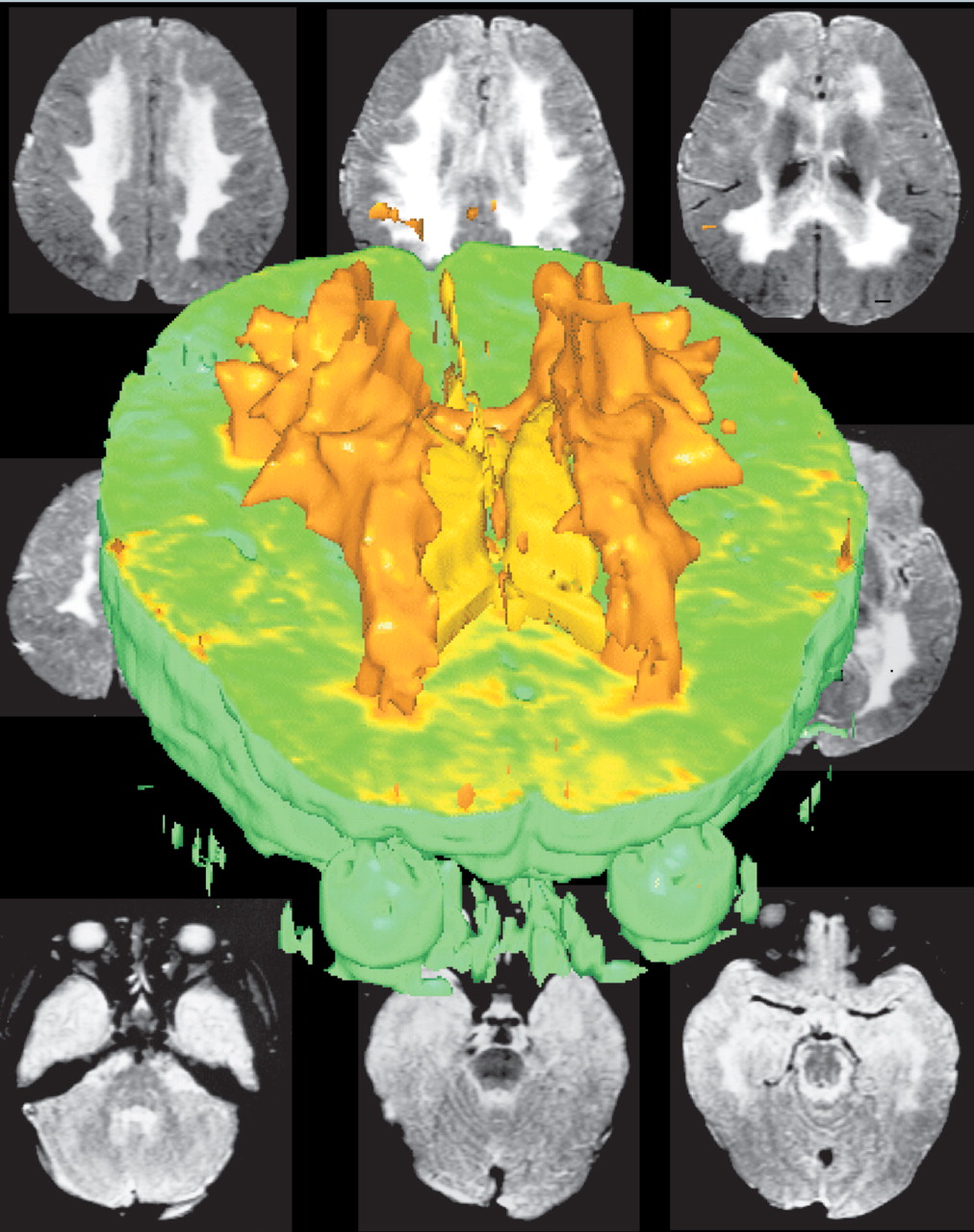
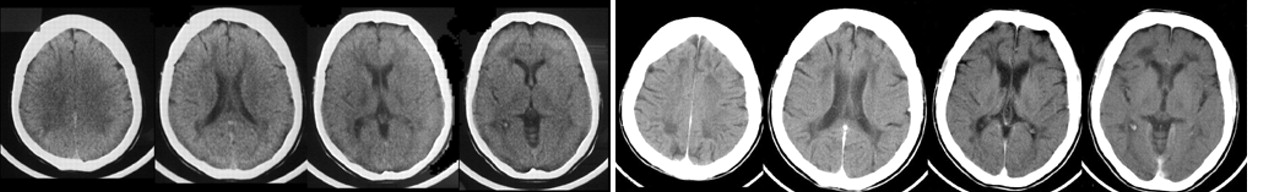
Computed tomography (CT) findings in MLD consist of confluent attenuation of signal from hemispheric white matter (
Figure 1). This is particularly marked in frontal and parietal regions. Later signs include progressive cortical and subcortical atrophy and ventricular dilatation
ex vacuo.9,12 White matter hypodensities on CT probably reflect myelin loss rather than pathological accumulation of cerebroside.
12Magnetic resonance imaging (MRI) is more sensitive than CT for the detection of white matter lesions of the brain (
Figure 2).
13 MRI is particularly useful for the visualization of early lesions, involvement of posterior fossa structures (brainstem and cerebellum) and for evaluating the severity and extent of the disease.
14 On T1 (inversion recovery)-weighted sequences, brain white matter has high signal, which reflects the presence of free water in the 150 nm spaces between the myelin lamellae. In contrast, brain white matter has low signal and cerebrospinal fluid (CSF) is hyperintense on T2-weighted sequences, which are more useful than T1 sequences for the detection of lesions of myelin.
13 FLAIR (fluid-attenuated inversion recovery) sequences produce T2 weighted images with the additional advantage of suppressing the CSF signal, facilitating the distinction between white matter lesions and enlarged Virchow-Robin spaces and lacunes.
13Like other leukodystrophies, MLD is distinguished from the asymmetrical white matter changes of normal aging, small-vessel disease, and multiple sclerosis by relatively symmetrical and confluent involvement of the hemispheric white matter. Most cases show lack of enhancement after contrast agent administration, although Kim et al.
15 reported linear enhancement of radial bands of intact perivascular myelin alternating with affected myelin in a tigroid or spotty pattern in 6 affected children, a pattern similar to Pelizaeus-Merzbacher leukodystrophy.
15 Progressive demyelination is believed to spread anteriorly to posteriorly, although exceptions to this pattern may occur.
15 Subcortical U-fibers are initially spared.
15 MLD has the added distinction of early and extensive involvement of the corpus callosum, internal capsules, corticospinal tracts in the brainstem, and deep cerebellar white matter.
13,15 Abnormal low signal in the thalami on T2-weighted images has also been reported.
14,15 Despite these features, T2 weighted MRI alone cannot definitively distinguish MLD from other dysmyelinating leukodystrophies, such as adrenoleukodystrophy and Krabbe's (globoid cell) leukodystrophy.
14Diffusion weighted MR (DWI), which is sensitive to the mobility of water in tissue, may be more specific than standard T2-weighted MRI in distinguishing MLD from other leukodystrophies, although there are few studies at present.
16,17 The apparent diffusion constant (ADC) was reduced in the deep white matter (indicating more restricted diffusion) of an infant with MLD on images acquired at 10 and 16 months of age. Restriction of diffusion in the diseased white matter was similar to the changes seen in cytotoxic edema.
17 Reduced ADC within the corpus callosum and inner portions of the centrum semiovale was also found in a 4½ year-old child with MLD.
16 The authors suggest that the accumulation of lysosomal membrane-bound sulfatides may restrict the movement of water in affected tissue.
16 In addition, directionality of diffusion, which is normally high in white matter, was also very low. The authors suggest that this may result from the extremely restricted diffusion in the affected white matter. These results are in contrast to most other leukodystrophies examined in this study, in which the ADC was increased, indicating less restricted diffusion.
In late-onset MLD, where the clinical presentation may resemble a primary psychiatric disorder, MRI is particularly useful in indicating the presence of white matter disease; indeed, pathological involvement of white matter may be seen on MRI before the onset of typical clinical signs.
12 MRI may, therefore, be a useful adjunct to biochemical screening for early disease detection in affected families.
14 Serial MRI may be a useful adjunct to clinical neurobehavioral evaluation to document decrease or stabilization of white matter pathology following therapeutic bone marrow transplantation.
18FUNCTIONAL IMAGING
Salmon et al.
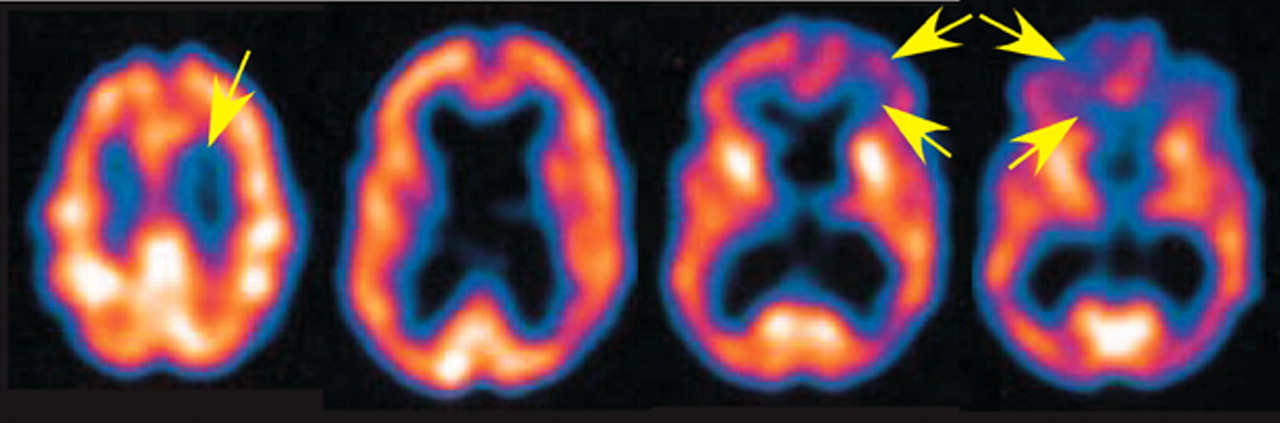
9 obtained positron emission tomography (PET) in a woman with enzymatically confirmed MLD who presented with cognitive changes in her mid-30s.
9 In contrast to the subcortical involvement of white matter on CT and MRI, hypometabolism was observed bilaterally in the thalamus, medial frontal cortex, frontal poles, and occipital cortex. This pattern differed from the characteristic dorsolateral prefrontal, parietal and posterior temporal association cortical hypometabolism in Alzheimer's dementia (AD). Tamagaki et al.
19 reported similar lack of correlation between T2 weighted MRI subcortical abnormalities and cortical hypoperfusion on SPECT in a patient with adult-onset MLD who presented behavioral abnormalities, euphoria, personality changes, and dementia.
19 Widespread functionally interconnected cortical regions may be disconnected by subcortical lesions. The clinical repercussions may resemble a primary cortical lesion (diaschisis) (
Figure 3).
A MODEL OF PSYCHOSIS
Late-onset MLD has been proposed as a naturally occurring model of schizophrenia.
2,20 The psychotic and cognitive features of late-onset MLD are extremely similar to those of schizophrenia. Both disorders are characterized by widespread anatomic dysconnectivity and secondary functional disruption. The prominent executive dysfunction in both MLD and schizophrenia reflects extensive disruption of frontal-subcortical circuits, which subserve mood, motivation, behavioral regulation, planning, and judgment.
21 Although abundant evidence suggests that schizophrenia is predominantly a disease of gray matter and MLD is a disease of white matter, there is evidence of neuronal dysfunction in MLD and white matter abnormalities in schizophrenia.
22–24 Decreased fractional anisotropy in the inferior frontal white matter is inversely correlated with negative symptoms in schizophrenia.
25 Furthermore, the anatomical distinction between cortical and subcortical disease is not corroborated by either clinical observation or functional imaging. A functional disturbance of neuronal metabolism and activity at a remote site after injury to an anatomically connected area of brain (diaschisis) is frequently found, and it may be the explanation for the SPECT observation of thalamic and widespread cortical hypoperfusion in the adult case of MLD, above.
9,19Some refractory patients with schizophrenia have been shown to have ARS-A pseudodeficiency or intermediate ARS-A levels between normal and deficient.
8 This supports the suggestion that accumulation of cerebroside sulfate is toxic to the brain. However, these observations have not been consistently replicated, in part because of differing assay techniques, widely variable normal ARS-A levels, and poorly controlled case definition. Additionally, it has not been definitively established that ARS-A deficiency or pseudodeficiency is higher in the psychiatric population than in the general population.
5,8,26,27 One interpretation of these discrepant results, based on
in vitro studies, is that low-enzyme activity individuals may be at greater risk of neuropsychiatric disability if they are exposed to environmental or endogenous stressors such as ethanol consumption.
8,11 An additional interpretation of these discrepant findings is that there may be a subgroup of patients with schizophrenia who have low ARS-A levels. These could be obscured by mean group comparisons.
28Evidence for the importance of white matter pathology in both schizophrenia and MLD is compelling. It has been proposed that schizophrenia is characterized by a developmentally mediated abnormal connectivity (“dysconnectivity”) between the prefrontal and mesiotemporal cortex.
29,30 In this model, subtle disorganization of cortical cytoarchitecture leads to disruption of neuronal circuits in widely distributed, functionally interrelated brain regions.
31 Loss of the normally dense and reciprocal projections between the prefrontal and limbic cortex is supported by anatomical studies showing reversal of the normal positive correlation between the volumes of left prefrontal cortex, superior temporal cortex, and hippocampus in patients with schizophrenia.
32 Functional imaging studies suggest that hypoactivation of prefrontal cortex is associated with dysregulation of mesiotemporal and ventral striatal regions, overactivity of limbic dopaminergic systems, and the “positive” manifestations of psychosis, namely, hallucinations and delusions.
33,34 Secondary disruption of white matter tracts is suggested by the finding of reduced volume of the mid-corpus callosum, which conveys axonal projections between the temporal lobes.
35 Some diffusion tensor imaging studies in schizophrenics show lower diffusion anisotropy in the prefrontal white matter and in the splenium of the corpus callosum relative to healthy controls (but cf).
36–39 Disruption of white matter may be secondary to cortical neuronal dysfunctio, or a primary deficit in myelin gene expression in oligodendroglia.
13,40,41CONCLUSION
There are several parallels between MLD and schizophrenia that support the value of MLD as a model for psychosis. To date, it is not clear whether MLD resembles schizophrenia because of widespread cortical and subcortical disconnection in both diseases or whether specific brain sites must be involved to cause the clinical phenotype of schizophrenia, independent of pathology. What is needed are comparisons of the areas of anatomical overlap between MLD and schizophrenia to define the necessary common ground for clinical disease. Suzuki et al., using three-dimensional MRI in a group of patients with schizophrenia, reported significant white matter decrease in the anterior limbs of the internal capsules and superior occipitofrontal fasciculus.
42 In contrast, Kim et al.
15 described white matter changes in the posterior limb of the internal capsule in a group of seven children with infantile MLD.
15 The anterior limbs were spared in all cases. The anterior limb of the internal capsule conveys reciprocal connections between the dorsomedian thalamus and the prefrontal cortex, while the posterior limb of the internal capsule conveys mostly corticospinal and corticobulbar motor fibers. The schizophreniform psychosis in adult MLD may reflect involvement of the thalamus in this condition, with secondary repercussions in the prefrontal cortex.
9 Further careful anatomical and functional neuroimaging studies will be useful in defining the anatomical substrate for diverse neuropsychiatric disorders with overlapping clinical manifestations.