Valproate was first approved in the United States in 1978 as an immediate-release formulation (Depakene) for the treatment of absence seizures. In 1983 another formulation, divalproex sodium (Depakote), was introduced, which is an enteric-coated stable coordination complex of valproic acid and valproate sodium.
Metabolized in the gut to valproate, divalproex sodium was designed to decrease the rate of absorption, thereby minimizing gastrointestinal side effects related to peak serum concentration. Although gastrointestinal discomfort may be reduced if valproate therapy is initiated with divalproex (
1), side effects are usually transient (
2) and may subside by four weeks of use (
3). A generic formulation of Depakene has been available since the mid-1980s and is bioequivalent to the brand-name product (
4).
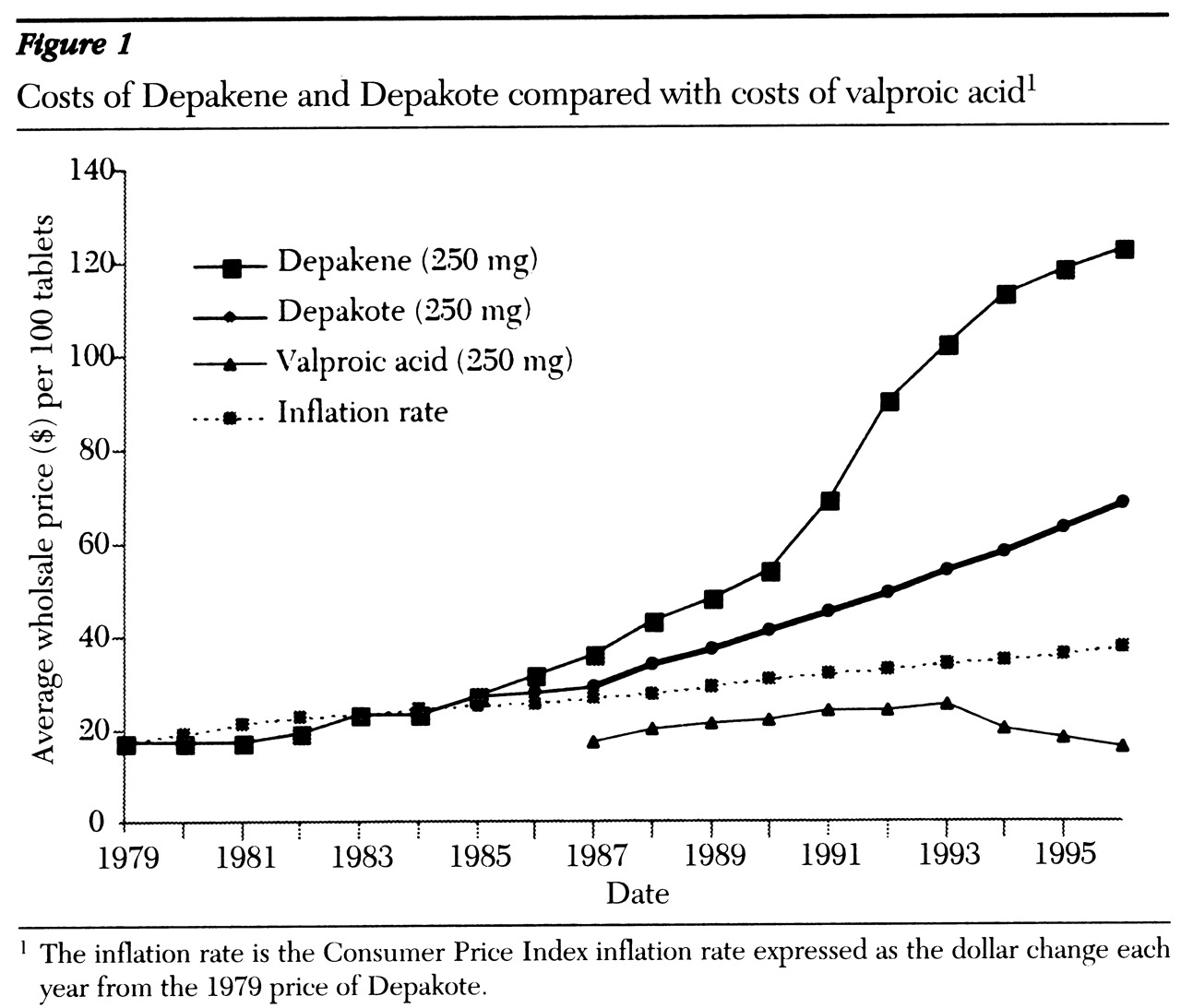
Over the years, the use of valproate has expanded to include treatment of complex partial and other seizures, the initial treatment of mania, migraine headache prophylaxis, and treatment of symptoms of behavioral dyscontrol. Drug costs became an issue as the prices of Depakene and Depakote rose substantially compared with the price of the generic valproic acid (
5) (see
Figure 1). This combination of widespread use and exorbitant costs prompted our examination of therapeutic substitution. Therefore, we prospectively studied the efficacy and tolerability of substituting valproic acid for divalproex in an adult psychiatric population stabilized on divalproex. The economic implications of the substitution also were examined.
Methods
This study was conducted at a state psychiatric hospital during 1996-1997. Subjects had been receiving a stable dose of divalproex for more than four weeks, were over 18, and were residing on chronic care wards. Patients were excluded if they had a history of intolerance to immediate-release valproic acid or if they were psychiatrically or neurologically unstable.
The substitution and monitoring procedures for the dosage form were initiated with the advice and consent of the hospital pharmacy and therapeutics committee as a component of formulary management. Patients were switched from divalproex to valproic acid at the same dose and interval using an open-trial design. After the switch, tolerability and efficacy were monitored and evaluated over a two-week period and again at six months.
A checklist of valproate side effects was developed to assess nausea, vomiting, diarrhea, constipation, indigestion, abdominal pain or cramps, changes in appetite, sedation-lethargy, and dizziness-ataxia. Patient responses were evaluated daily and recorded as mild, moderate, severe, or not noted. The Clinical Global Impressions scale (CGI) was completed weekly for each patient by team consensus. Seizure frequency was measured by event occurrence. Morning trough serum concentrations of valproate were drawn two weeks after switching. CGI change scores were analyzed using one-sample t tests. Serum valproate concentrations and CGI severity scores from baseline to six months were analyzed using two-tailed paired t tests.
Results
The 47 subjects in the study had a mean±SD age of 38.9±11.3 years (range, 18 to 73 years). Twenty-five (53 percent) were Caucasians, and 20 (43 percent) were African Americans. Twenty-nine (62 percent) were male. Indications for valproate included mood disorder (bipolar disorder and schizoaffective disorder) for 25 patients, behavioral dyscontrol (aggression, assaultiveness, impulsiveness, and combativeness) for 20 patients, and seizures (myoclonic jerks, tonic-clonic seizures, and complex partial seizures) for seven patients.
Forty-six patients (98 percent) completed the two-week study. For one patient, valproic acid was discontinued for reasons unrelated to the substitution. The mean±SD dose of valproate remained unchanged at 1,505.3±604.5 mg a day. Dosing intervals remained unaltered for all but two patients, whose single doses of divalproex greater than two grams were a priori divided into twice-daily dosing.
No change in seizure occurrence or CGI scores was noted. The mean baseline CGI severity measurement was 4.44 (range=3 to 6); on the CGI, 4 indicates moderate severity and 5 marked severity. CGI change scores reflect change from baseline, with 4 representing no change, 3 minimal improvement, and 5 minimal worsening. The one-week mean CGI change score was 4.02 (range, 4 to 5). At two weeks, the mean CGI change score was 4.08 (range, 3 to 6).
Mean±SD baseline and two-week trough concentrations were 69.14± 21.35 μg/ml and 59.21±14.66 μg/ml, respectively. The decrease of -9.93±17.75 μg/ml was statistically significant (t=3.46, df=41, p=.001; 95 percent confidence interval=-14.39 to -4.46; range=30.3 to -50.3 μg/ml).
Fourteen patients had gastrointestinal complaints during the month before the switch in medication. Five patients had gastrointestinal complaints during the study period. For four of the five patients, the complaints were mild to moderate and transient. Only one patient was assessed as experiencing persistent moderate gastrointestinal side effects resulting in discontinuation of valproate and reinstitution of divalproex.
At the six-month follow-up, efficacy and tolerability were reevaluated. Of the initial 47 patients, 25 had been discharged, and 19 remained on valproic acid treatment. For three patients switched back to divalproex, gastrointestinal complaints were not clearly linked to the substitution and continued after reinstitution of divalproex.
Among those remaining hospitalized, the mean baseline CGI severity scores was 4.76 (range=3 to 6), and at six months it was 4.41 (range=3 to 6). Although the CGI scores showed a statistical trend toward improvement (p=.055; CI=-.714 to .008), no evidence was found of any change related solely to the substitution, nor were any additional cases of intolerable gastrointestinal side effects noted.
Discussion and conclusions
In this study, all patients but one were able to successfully tolerate the therapeutic substitution of the immediate-release product for the enteric-coated formulation. These patients were allowed time to gain tolerance to gastrointestinal irritation by beginning treatment with the enteric-coated product before substitution with valproic acid. Only five of 47 patients (11 percent) experienced gastrointestinal complaints during the study period. For only one patient (2 percent) were the complaints persistent and related to the valproate, requiring reinstitution of divalproex sodium.
The mean 12-hour trough serum concentrations of valproate decreased by 14.4 percent, which has been observed by others (
6,
7). Although a statistically significant decrease occurred, no evidence that this change was clinically relevant was found. Efficacy measurements remained unaltered, and mean serum concentrations remained within the therapeutic range (45 to 125 μg/ml).
Based on these results, it is apparent that converting patients from divalproex to valproic acid could be a direct means of cost savings. Acquisition costs for 1,500 mg a day for divalproex tablets is $3 per patient per day versus $.52 per patient per day (including costs for in-house unit-dose packaging) for valproic acid. Thus medication costs were reduced 83 percent, or approximately $905 per patient-year.
This preliminary study has several methodological limitations. First, the substitution was not double-blinded, and the premise of the substitution included a bias to support the valproate. The lack of a control group makes it difficult to draw firm conclusions about the incidence of minor side effects. However, we actively sought patient complaints of side effects each day. Our side effect rate of 11 percent and discontinuation rate of 2 percent are consistent with those in previous reports (
8).
Second, valproate was being used for multiple indications in this diagnostically heterogeneous sample. Thus, although the CGI was used as a global measure, it may not have been adequate to assess the range of specific clinical changes that might occur. Also, although the same raters followed individual patients, no interrater reliability testing was done. No clinical or statistically significant changes in symptom severity were found; however, more sensitive and specific ratings may have revealed changes.
Finally, the sample size may not have been large enough to demonstrate a difference in adverse effects following substitution. Power analysis suggests that if the incidence of gastrointestinal intolerance is 2 percent, a cohort of 300 patients would be needed to demonstrate a difference in gastrointestinal side effects at p<.05.
Nonetheless, to our knowledge this study offers the first prospective report of the substitution of valproic acid for divalproex. Our findings replicate the retrospective reports of Vadney and associates (
9) and Cranor and colleagues (
10) of substitution of valproic acid among persons with developmental disabilities.
These preliminary results suggest that many chronic psychiatric inpatients stabilized on divalproex may be safely switched to valproic acid. Future investigations should focus on replicating these findings in a larger population. The applicability of these findings to other settings, such as outpatient care, or for other indications, such as migraine headache prophylaxis, needs to be determined.
Acknowledgments
The authors thank Raymond C. Love, Pharm.D., Aaron Burstein, Pharm.D., Mary Borovicka, Pharm.D., and David Moore, R.Ph., M.P.A., for their review and comments.