Synaptic pathology has received increasing interest as a key feature of schizophrenia’s neuropathology
(1–
3) and possibly its pathogenesis
(4–
10) and genetic etiology
(11). Most investigations of synaptic pathology in schizophrenia have used presynaptic proteins and their mRNAs as molecular markers, respectively providing information about the synaptic terminals and the neurons that give rise to them
(12). Particularly in the hippocampal formation, there is relatively consistent evidence for low expression of genes for presynaptic proteins, such as synaptophysin
(13–
17). The precise anatomical distribution and neurochemical phenotype of the affected synapses remain largely unknown, although glutamatergic synapses may be preferentially involved
(18–
20), and the CA1 subfield may be spared more than the rest of Ammon’s horn and the subiculum
(13,
15,
16).
The preceding data suggest that aberrant synaptic connectivity may contribute to hippocampal dysfunction in schizophrenia
(21,
22). However, such inferences are premature without information about the status of the postsynaptic elements of the synapses. Hence, evaluation of hippocampal dendrites and dendritic spines (the protuberances on which most excitatory synapses are apposed) is necessary. To date, the only data of which we are aware are reports of decreased dendritic arborization and spine density in subicular pyramidal neurons
(23) and reduced immunoreactivity of microtubule-associated protein 2 (MAP2), a dendritic protein, in some
(24,
25) but not all
(26,
27) studies. Thus, we examined the expression of genes for two dendritic proteins: MAP2 and spinophilin (neurabin II), a marker of the dendritic spine
(28,
29). We used in situ hybridization, to allow the neurons expressing these genes to be studied and to be able to relate the subfield distribution of abnormalities to the reported presynaptic protein findings. We included a group of subjects with mood disorders, for which there is preliminary evidence of presynaptic
(16,
30,
31) and dendritic
(23) pathology.
Discussion
Hypotheses that aberrant synaptic development and plasticity are important in schizophrenia
(4–
6,
9) are increasingly being supported by convergent empirical data from molecular and neuropathological studies
(1,
3,
8,
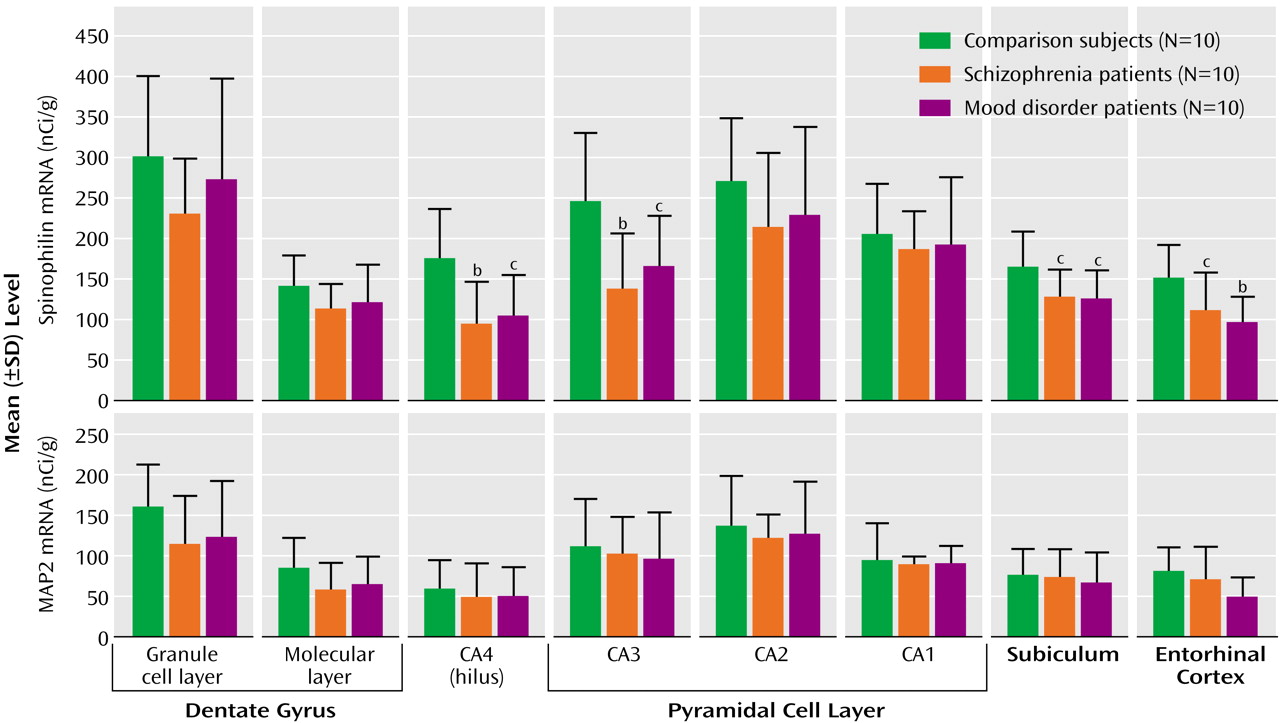
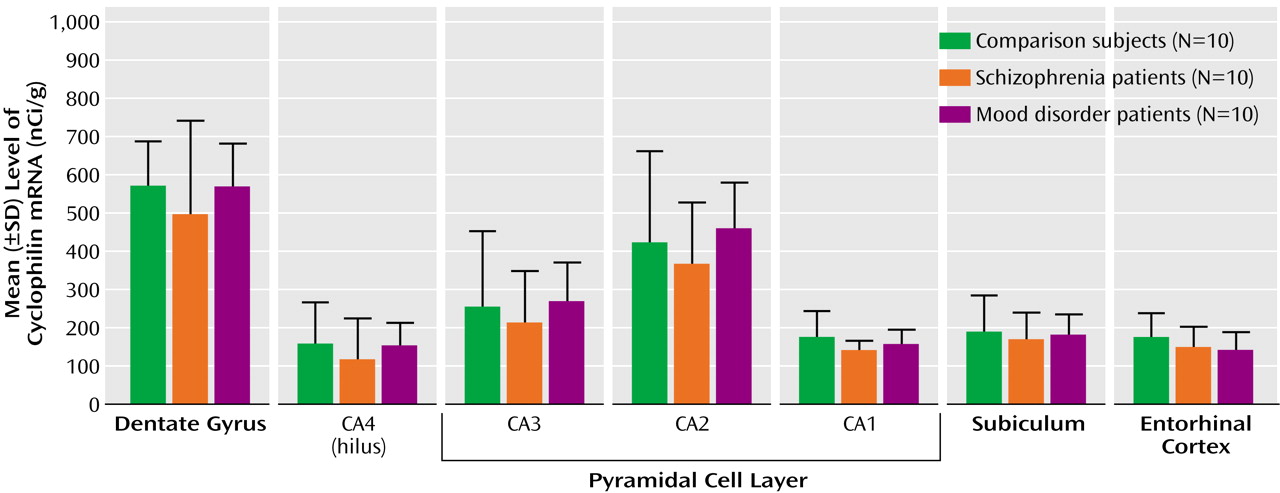
17). However, most of the existing data regarding the hippocampal formation pertain to the presynaptic side of the synapse, and little is known about the key postsynaptic elements: dendrites and their spines. This study provides molecular evidence that the latter are also affected in schizophrenia, with a lower level of the mRNA encoding the dendritic spine protein spinophilin. This deficit was molecularly specific, in that no abnormality occurred in transcripts encoding the dendritic marker MAP2 and the housekeeping gene cyclophilin.
The simplest interpretation of the spinophilin reduction is that it reflects a loss of dendritic spines. This morphological interpretation is consistent with the findings of Rosoklija et al.
(23) in the subiculum and suggests that similar changes occur in several other hippocampal subfields. It is also in keeping with experimental studies showing that alterations in dendritic spine number are accompanied by parallel changes in spinophilin expression
(28,
40). A similar relationship pertains between MAP2 expression and overall dendritic extent
(41–
43); hence, our negative results for MAP2 suggest that the total dendritic tree of hippocampal neurons is not markedly altered in schizophrenia. An alternative interpretation of the reduced spinophilin gene expression in schizophrenia is that it reflects a decreased activity or plasticity of dendritic spines, rather than (or as well as) morphological change per se
(7). This interpretation is plausible since spines are dynamic structures central to synaptic plasticity and affected by synaptic activity
(44,
45) and spinophilin is implicated in these processes
(26,
46,
47).
The deficits in hippocampal expression of genes for presynaptic proteins in this
(16) and other
(13,
15,
17,
30) groups of schizophrenia patients and the positive correlations between synaptophysin mRNA and spinophilin mRNA, in our total study group and in the disease groups separately, raise the question of how these facets of synaptic pathology are related. Regarding the direction of causality, precedents suggest that dendritic pathology is likely to be secondary to abnormal afferent innervation, rather than the other way around
(48–
51). However, consideration of the anatomical correspondence between pre- and postsynaptic abnormalities suggests that the relationship may be indirect: the synaptophysin mRNA studies
(13,
15,
16) all show substantially lower levels in CA3, which provides the primary input to CA1 neurons via the Schaffer collaterals, but no difference in CA1. Hence, if there were a direct relationship between pre- and postsynaptic changes, spinophilin mRNA would likely be decreased in CA1 to accompany the loss of synaptophysin mRNA in CA3. In fact, spinophilin mRNA showed a subfield profile of abnormalities similar to that of synaptophysin mRNA, with CA1 again unaffected. Thus, an alternative explanation is that there are intrinsic differences in the vulnerability of CA3 and CA1 neurons to synaptic pathology, with those in CA1 being resistant to both presynaptic and dendritic involvement. One determinant of this differential vulnerability may be genetic, in that three of the putative schizophrenia susceptibility genes, neuregulin, dysbindin, and calcineurin gamma, show much lower expression in CA1 than in the rest of the hippocampal formation (unpublished observations). It may also be that, in schizophrenia, other CA1 afferents (e.g., commissural or monoaminergic) are unusually high and compensate for the low input from CA3 or that CA3 neurons are intrinsically more sensitive to fluctuations in gene expression.
Most glutamatergic (excitatory) synapses terminate on dendritic spines, and most axospinous synapses are glutamatergic. Hence, the present results support the view that, at least within the hippocampal formation, glutamatergic neurotransmission is particularly affected in schizophrenia
(19,
52,
53). An interesting development is the recent report that the abundance of GluR2, a key non-
N-methyl-
d-aspartate receptor subunit, regulates dendritic spine formation in hippocampal neurons
(54). Since GluR2 expression is reduced in hippocampal neurons of schizophrenia patients
(55,
56), GluR2 may be part of the molecular cascade that leads to the dendritic spine pathology of schizophrenia. This correspondence of findings also emphasizes that, at the molecular level, there is a continuum, not a distinction, between structural and functional (neurochemical) aspects of neuropathology.
Reductions in spinophilin mRNA were also observed in the mood disorder group, consistent with initial morphological evidence for reduced dendritic spine density
(23). This suggests considerable overlap between schizophrenia and mood disorders regarding involvement of dendritic spines. Whether this similarity of pathology reflects similar pathogenic processes, such as those already outlined, remains to be determined.
Several potential limitations of this study should be noted. First, the small number of subjects with bipolar disorder required us to group them with the subjects who had major depression in a single mood disorder group for the analyses. The possibility of different abnormalities of dendritic gene expression among mood disorder subtypes thus requires further study; however, inspection of our data did not provide any indication of this.
Second, autoradiographic film analyses can be confounded by differences in the packing density of neurons between the groups being compared. Although there is no good evidence that this occurs in the hippocampus in schizophrenia
(1) or mood disorders
(31), we investigated the possibility directly by doing a “per cell” analysis of spinophilin mRNA in CA4 and CA1 neurons, using the emulsion-dipped sections. This showed that spinophilin mRNA is lower in CA4 neurons but not in CA1 neurons in schizophrenia (data not shown), confirming the film results.
Third, medication might have contributed to, or masked, differences between diagnostic groups. However, there were no correlations of the mRNAs with either lifetime or recent neuroleptic exposure, and chronic haloperidol and chlorpromazine treatment have no effect on spinophilin mRNA in the rat hippocampus
(36). It also seems unlikely that occult substance misuse among the patients confounded the results, since the patients had negative toxicology results and since psychostimulants increase, not decrease, dendritic spines
(57). A similar argument pertains to the possible effect of antidepressants and mood stabilizers, which increase spine densities in antidepressant-treated animals
(31,
58).
Fourth, we found correlations of spinophilin mRNA with brain pH and, less clearly, with postmortem interval. However, these variables did not differ significantly between groups, and all the positive results remained significant when pH and postmortem interval were included as covariates in an analysis of covariance (data not shown).
Finally, a different limitation comes from the fact that we measured only the mRNAs and not the proteins. This decision was made because of the existence of multiple isoforms and phosphorylation states, which complicates quantitative antibody-based methods
(26,
27), because the proteins may be sensitive to perimortem degradation
(59), and also because of limited tissue availability. It is reassuring that prior studies have shown a good correspondence between protein and mRNA levels for several dendritic proteins
(42,
60,
61). Nevertheless, evaluation of both types of gene product will be desirable in future work, which is also needed to confirm our observations and extend them to other brain regions wherein dendritic pathology has been reported in schizophrenia
(62–
64).
In conclusion, our results provide molecular evidence for a postsynaptic component of glutamatergic synaptic pathology in the hippocampal formation in schizophrenia and mood disorders. Decreased spinophilin expression suggests that the structural and functional integrity of dendritic spines is compromised. The combination of presynaptic and postsynaptic abnormalities may represent an important component of these disorders.