The chronicity of schizophrenia determines the primary rationale for studies of early detection and early intervention for this disorder. A preventive approach was suggested many years ago
(1), but not until developments in the last decade has it proven feasible, ethical, and attractive from a risk-benefit perspective
(2) . These developments include the introduction of atypical antipsychotics and the ability to identify with reasonable psychometric precision, reliability, and predictive validity persons at high risk for psychosis in the early (prodromal) phases of schizophrenia
(3) .
Despite the importance of prodromal intervention studies in schizophrenia, to our knowledge only three randomized studies have been conducted to date
(3 –
7) . The first study, by McGorry et al.
(4), was a randomized clinical trial of patients meeting criteria for high risk and prodromal symptoms
(8,
9) and provides the strongest evidence to date of the efficacy and safety of early intervention. In addition to the needs-based intervention, the experimental group in this study received open-label risperidone (1–2 mg/day) plus cognitive behavior stress management for 6 months, followed by 6 months of no treatment. The control group received needs-based intervention only. The second study, a randomized trial of cognitive behavior therapy conducted by Morrison et al.
(5), suggests that psychosocial treatment alone may prevent or delay the onset of schizophrenia. The third study, presented in the current report, is to our knowledge the first double-blind, placebo-controlled clinical trial of an atypical antipsychotic, olanzapine, for patients with prodromal symptoms of schizophrenia at high risk for psychosis.
The study objectives were to determine whether olanzapine can delay or prevent the onset of psychosis and reduce prodromal symptoms with adequate safety. The current report focuses on the effects of olanzapine during the double-blind treatment year and the follow-up (no-treatment) year. Three previously published reports detail the study rationale and design
(3), the recruitment procedures and baseline characteristics of the prodromal study group
(6), and the efficacy and safety of olanzapine versus placebo in the first 8 weeks of the double-blind period
(7) .
Briefly, the study included help-seeking persons between ages 12 and 45 years who responded to advertisements or who were referred by clinicians. Persons meeting the study criteria for the prodrome were told that they had symptoms suggesting an increased risk of developing psychosis. They were informed of the study and invited to consent to participate. Excluded were persons who met the criteria for a past or current psychotic disorder, who had a treatable psychiatric disorder that could account for the prodromal symptoms, who were suicidal or homicidal, or whose prodromal symptoms were primarily sequelae of drug or alcohol use.
The study consisted of four periods: 1) 3- to 14-day screening, 2) a 1-year period of double-blind treatment with olanzapine (5–15 mg/day) or placebo, 3) a 1-year no-treatment follow-up period, and 4) a 6-month period of open-label treatment with olanzapine (5–10 mg/day) for patients who experienced a conversion to psychosis. This report focuses on study periods 2 (double-blind treatment) and 3 (no-treatment follow-up). In period 2, the study drug consisted of 5-mg olanzapine tablets or placebo tablets dispensed in blister packs that allowed monitoring of drug compliance. Concomitant psychoactive medications were not allowed, and supportive psychosocial interventions were available to all patients and their families.
Before being randomly assigned to a study drug, each patient signed an informed consent form. The study protocol was approved by the appropriate institutional review boards.
Method
This study was conducted from 1997 to 2003 at the Prevention Through Risk Identification, Management, and Education (PRIME) clinics at four sites: Yale University, New Haven, Conn. (T.H.M., primary investigator); University of Toronto, Toronto (R.B.Z., primary investigator); University of North Carolina, Chapel Hill (D.P., primary investigator); and University of Calgary, Calgary, Alta., Canada (J.A., primary investigator).
In this article, “the prodrome to onset of psychosis” refers to a virtual syndrome that is validated only after the onset; thus, the term “prodromal” here means “putatively prodromal.” We used the Structured Interview for Prodromal Syndromes
(10,
11) to determine whether the patients met the Criteria for Prodromal Syndromes
(12) . In turn, the Criteria for Prodromal Syndromes (detailed in references
3,
12, and
13 ) operationally define the three prodromal syndromes outlined by Yung and McGorry
(8) : attenuated positive symptom syndrome, genetic risk and deterioration syndrome, and brief intermittent psychotic state syndrome.
Assessments
The primary efficacy measure for study periods 2 and 3 was the rate of conversion to psychosis. “Psychosis” was defined as psychotic disorders in the DSM-IV schizophrenia spectrum and was identified by using the Presence of Psychosis Scale
(3), which we developed because of the lack of DSM-IV-defined criteria for psychosis onset.
The secondary efficacy measures assessed changes in prodromal symptoms, schizophrenia symptoms, and patients’ functioning and included the Scale of Prodromal Symptoms
(10,
13), Positive and Negative Syndrome Scale (PANSS)
(14), Clinical Global Impression (CGI) severity of illness scale (
15, pp. 218–222), Montgomery-Åsberg Depression Rating Scale (MADRS)
(16), Young Mania Rating Scale
(17), and Global Assessment of Functioning Scale (GAF)
(18) . The Scale of Prodromal Symptoms, developed in 1997 by the senior author and colleagues, includes 19 items that assess positive symptoms, negative symptoms, disorganization symptoms, and general symptoms. All of the items are rated from 0 to 6: 0=not present, 3=moderate, 5=severe but not psychotic, 6=severe and psychotic. The Scale of Prodromal Symptoms is embedded within the Structured Interview for Prodromal Syndromes, which was designed to diagnose prodromal syndromes according to the criteria mentioned earlier
(3,
8,
13) and to rate the severity of prodromal symptoms.
Vital sign measurements, laboratory tests, and electrocardiograms were performed during both study periods. Extrapyramidal symptoms and abnormal involuntary movements were assessed during study period 2 by using the Simpson-Angus Rating Scale
(19), Abnormal Involuntary Movement Scale
(15, pp. 534–537), and Barnes Rating Scale for Drug-Induced Akathisia
(20) . Premorbid functioning was measured with the Cannon-Spoor Premorbid Adjustment Scale
(21), and family history of mental illness was assessed with the Modified Family History Research Diagnostic Criteria
(22) . Finally, each patient was thoroughly interviewed at the initial evaluation to determine the presence of current or past axis I and axis II disorders.
Statistical Analyses
All analyses were done on an intent-to-treat basis. All cited p values were two-sided with a significance level of 0.05. The planned primary analysis was to use the Fisher exact test to assess treatment differences in the conversion-to-psychosis rates during study periods 2 and 3 among those who completed the study or converted to psychosis. For the analyses in this report, the denominators for the conversion rates during study periods 2 and 3 are the total numbers of patients who entered these study periods. In retrospect, the Fisher exact test was not equipped to handle the varying durations of study participation that resulted from a high dropout rate and the variable timing of conversion to psychosis. A post hoc analysis of the Cox proportional hazards model of the data on time to onset of psychosis is included in this report to supplement the results from the primary analysis because this survival analysis was deemed more capable of handling right-censored observations, of accounting for the timing of the events, and of evaluating the primary efficacy measure. This Cox proportional hazards model included treatment and baseline scores for positive symptoms on the Scale of Prodromal Symptoms as the terms in the model. The score for prodromal positive symptoms was included in the model because positive symptoms as measured by the Scale of Prodromal Symptoms were used to define psychosis and conversion to psychosis.
Analysis of covariance (ANCOVA) was used to evaluate the changes in continuous efficacy variables during study period 2, based on the last observation carried forward. Analysis of variance (ANOVA) was used to evaluate such changes during study period 3 and changes in continuous safety variables during both study periods, with the last observation carried forward. ANCOVA models included terms for treatment group, investigator, and baseline value; the ANOVA models included terms for treatment group and investigator. For all analyses of continuous variables, last observation carried forward was used as the analytical protocol-defined method to handle missing data in the analyses of mean change. Since the development of this study protocol, this method has been documented to result in biased treatment estimates and their standard errors when the data violate the following assumptions: 1) data missing at random and 2) subject responses being constant from the last observed value to the endpoint of the trial. This bias can result in the inflation of type I error when the treatments do not show different outcomes and overestimation of the magnitude of treatment differences when the treatments do show different outcomes. For this reason, we employed an alternative analysis using a mixed-effects model repeated-measures ANCOVA to analyze the change from baseline to each postbaseline visit in the positive symptom score on the Scale of Prodromal Symptoms
(23) . A first-order autoregressive moving-average covariance matrix was determined by Akaike’s information criterion to be the best-fit covariance structure in modeling the within-patient error. The ANCOVA model included the categorical variables of investigator, treatment group, week of therapy, and interaction of treatment and week of therapy and the continuous covariate of baseline score as fixed effects.
Categorical data, which included demographic and illness characteristics, discontinuation rate, treatment-emergent adverse events, and treatment-emergent extrapyramidal symptoms, were evaluated by using Fisher’s exact test.
Results
Baseline Patient and Illness Characteristics
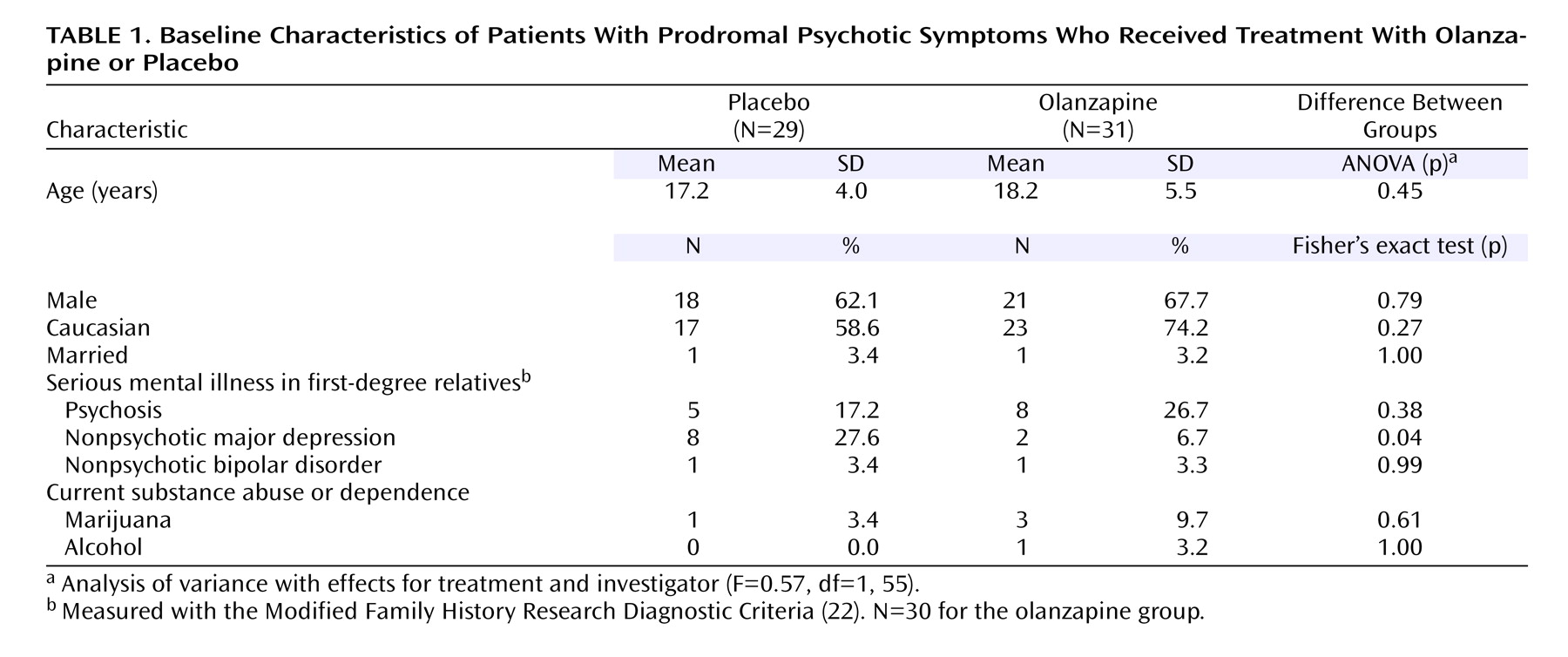
Sixty treatment-seeking patients with prodromal symptoms were recruited across the four sites. The overall study group was largely Caucasian (66.7%, N=40), young (median age=16 years, range=12–36), male (65.0%, N=39), and single (91.7%, N=55). Our second report
(6) details the overall patient characteristics.
Table 1 shows the distribution of demographic and illness characteristics in the two treatment groups. The only significant difference was in the rate of family history of nonpsychotic depression, which was higher in the placebo group. Compliance with the study drug regimen during study period 2 did not differ significantly between groups (data not shown).
Dropouts and Conversions to Psychosis
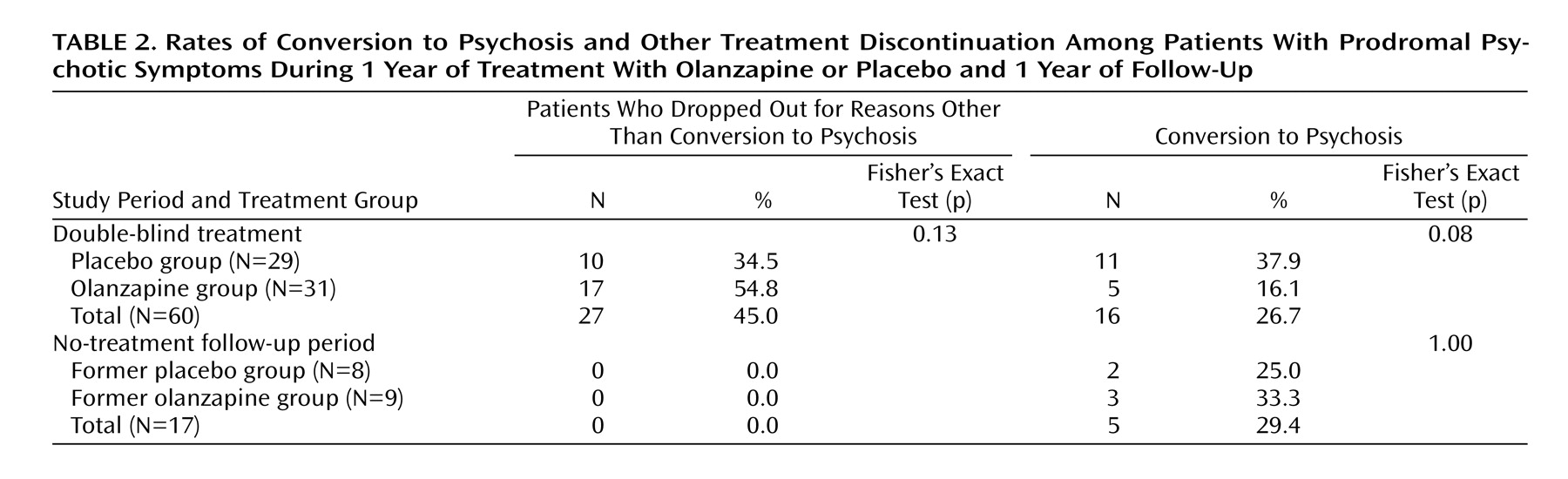
Table 2 presents the rates of patients who dropped out or whose symptoms converted to psychosis during each study period. During study period 2, the dropout rate for reasons other than conversion to psychosis was higher for the olanzapine group, but this did not differ significantly between treatment groups. In addition, no significant difference was observed in the rate of patients who discontinued this study period because of adverse events: 3.4% in the placebo group (one of 29) and 3.2% in the olanzapine group (one of 31).
During this period the rate of conversion to psychosis was higher for placebo, but the group difference was not significant (
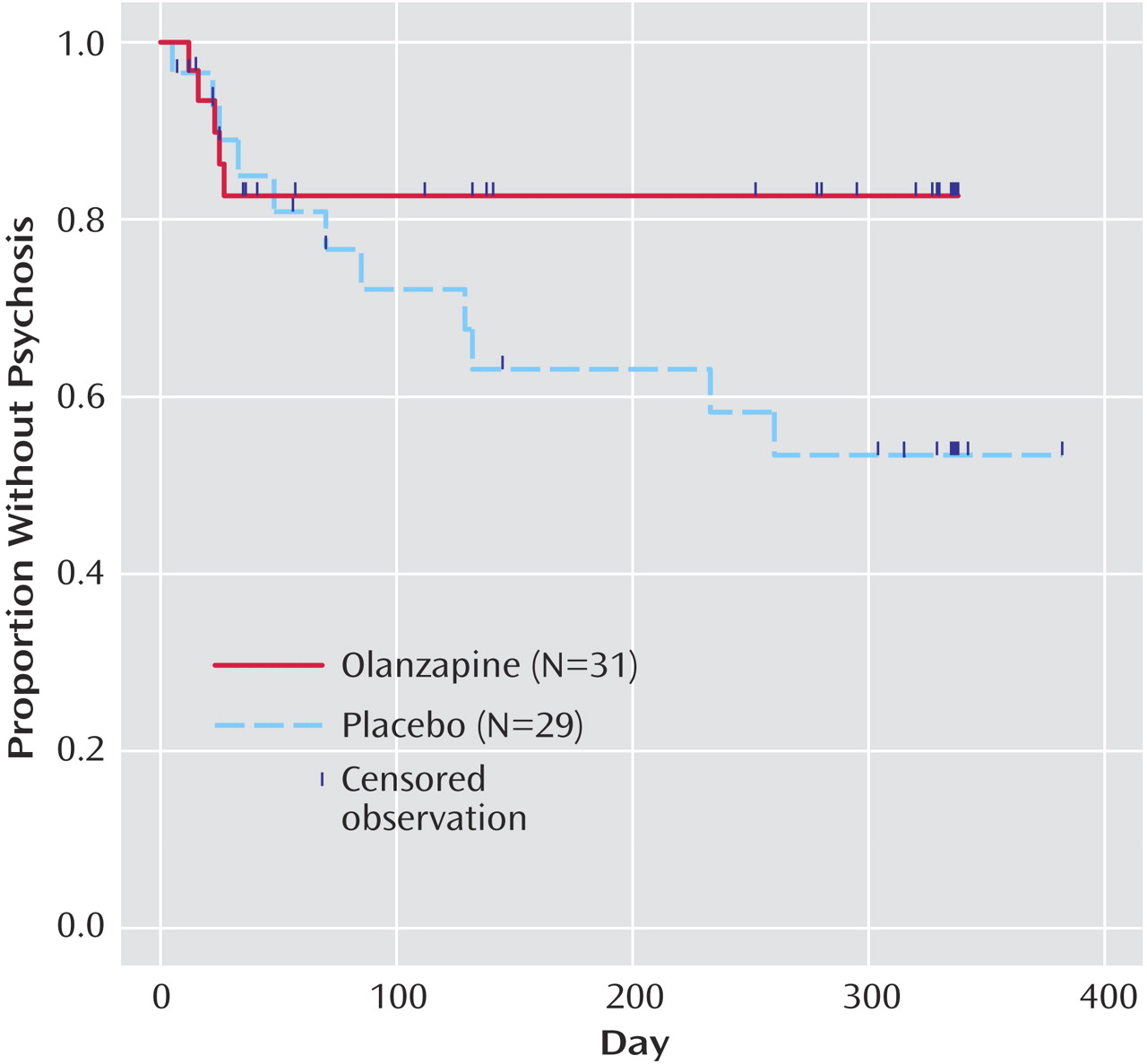
Table 2 ). All five of the olanzapine-treated patients who converted to psychosis did so within the first 4 weeks, whereas the 11 placebo-treated patients converted to psychosis throughout the entire year. Kaplan-Meier survival curves of the time to onset of psychosis during study period 2 are plotted in
Figure 1 . Results from the Cox proportional hazards model showed that the treatment groups differed to a nearly significant degree (p=0.09) in the hazard of converting to psychosis after adjustment for the baseline severity of positive prodromal symptoms: the hazard of conversion to psychosis for the placebo-treated patients was 2.5 times that for the olanzapine-treated patients.
Overall, 12 of the 60 patients (20.0%) completed both periods of the study. The overall rate of conversion to psychosis was 35.0% (21 of 60).
During study period 3, there were no discontinuations due to any reasons other than conversion to psychosis in either treatment group. Because of the low number of patients who entered this study period, no statistical analysis was performed to assess treatment differences in the time to onset of psychosis during this study period.
Efficacy for Prodromal Symptoms and Severity of Illness
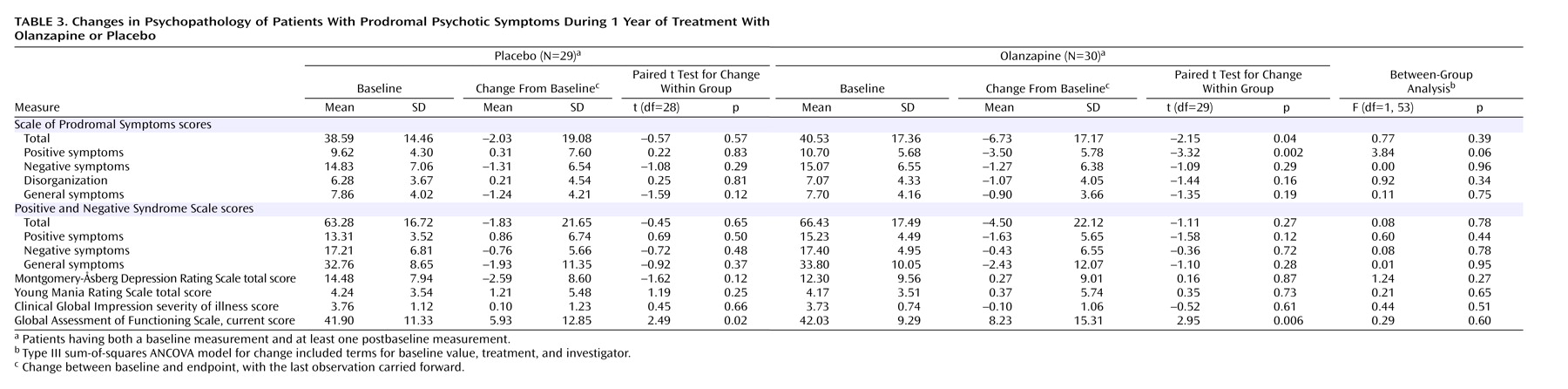
Table 3 shows patients’ changes in symptom and functioning scores during the double-blind treatment. The groups did not differ significantly in mean baseline-to-endpoint changes (last observation carried forward) in these scores. However, the olanzapine group showed a greater improvement in positive symptoms than the placebo group, and the difference approached significance. Significant baseline-to-endpoint changes were seen in the total score and positive symptom score on the Scale of Prodromal Symptoms within the olanzapine group and in the GAF current score within the olanzapine and placebo groups. The ratings of psychosis (PANSS), depression (MADRS), and mania (Young Mania Rating Scale) changed little in either group.
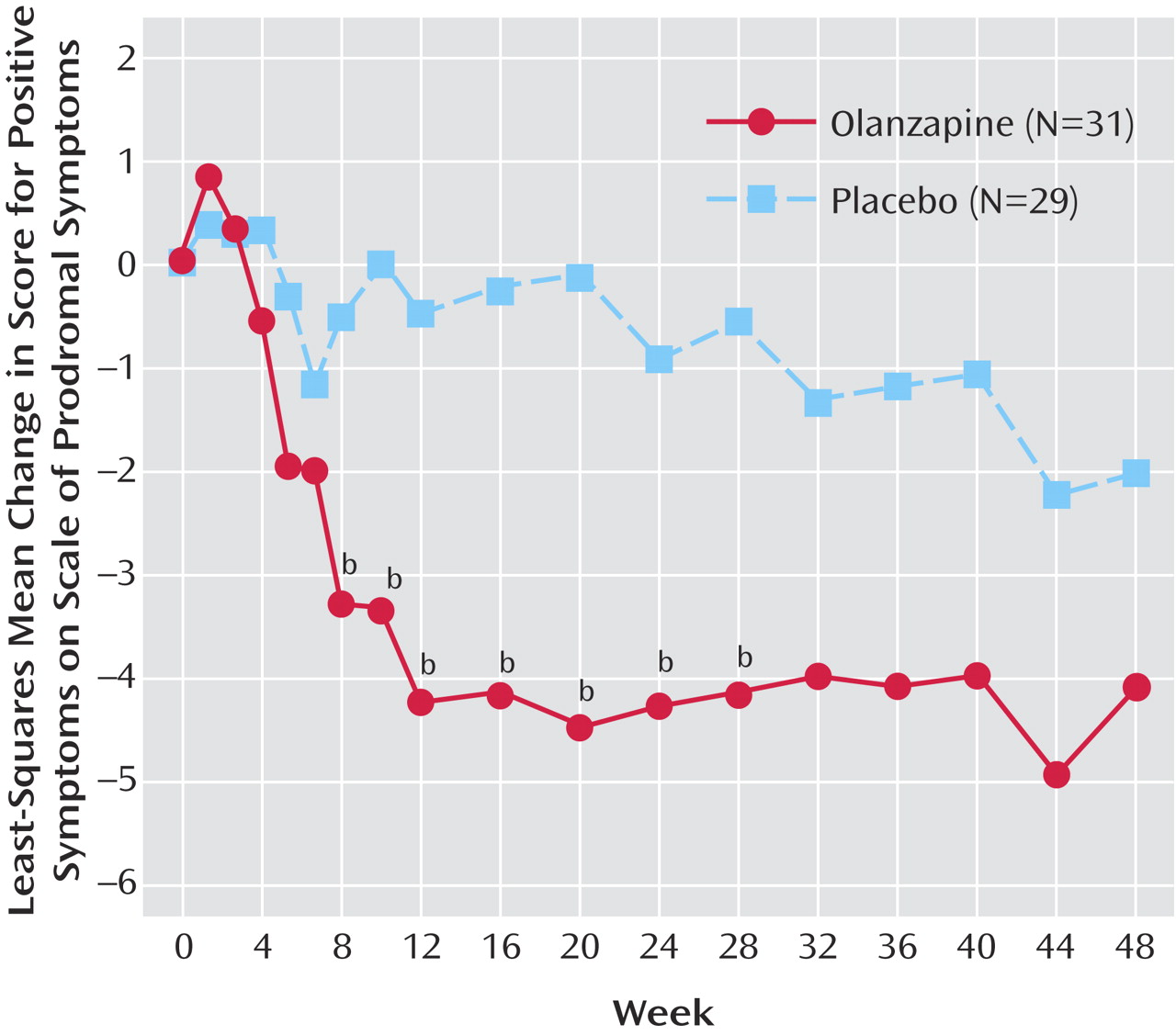
Figure 2 shows the mixed-effects model repeated-measures least-squares mean change in positive symptoms on the Scale of Prodromal Symptoms for the olanzapine and placebo groups at each postbaseline visit during study period 2. The overall treatment effect (F=5.21, df=1, 53, p<0.03) and the week-of-therapy effect (F=1.76, df=17, 555, p<0.03) were both significant, indicating that overall there were significant differences between treatment groups and that the positive symptom score significantly changed over time. The interaction of treatment and week of therapy was not significant (F=1.25, df=17, 555, p=0.22), indicating that the treatment differences did not significantly change over time. The only significant between-treatment differences were observed between weeks 8 and 28, when the least-squares mean changes in positive symptom scores were significantly greater for the olanzapine patients.
In study period 3 (the no-treatment follow-up), positive symptom scores on the Scale of Prodromal Symptoms worsened significantly for the nine patients from the former olanzapine group (baseline at end of treatment phase: mean=5.33, SD=5.43; change from baseline: mean=5.22, SD=5.85) (Wilcoxon signed-rank test, T=16.5, N=9, p=0.02). For the eight patients from the former placebo group, positive prodromal symptoms also worsened in this study period but not significantly (baseline for study period: mean=3.63, SD=3.93; change from baseline: mean=2.88, SD=8.61) (p=0.61, Wilcoxon signed-rank test).
Safety
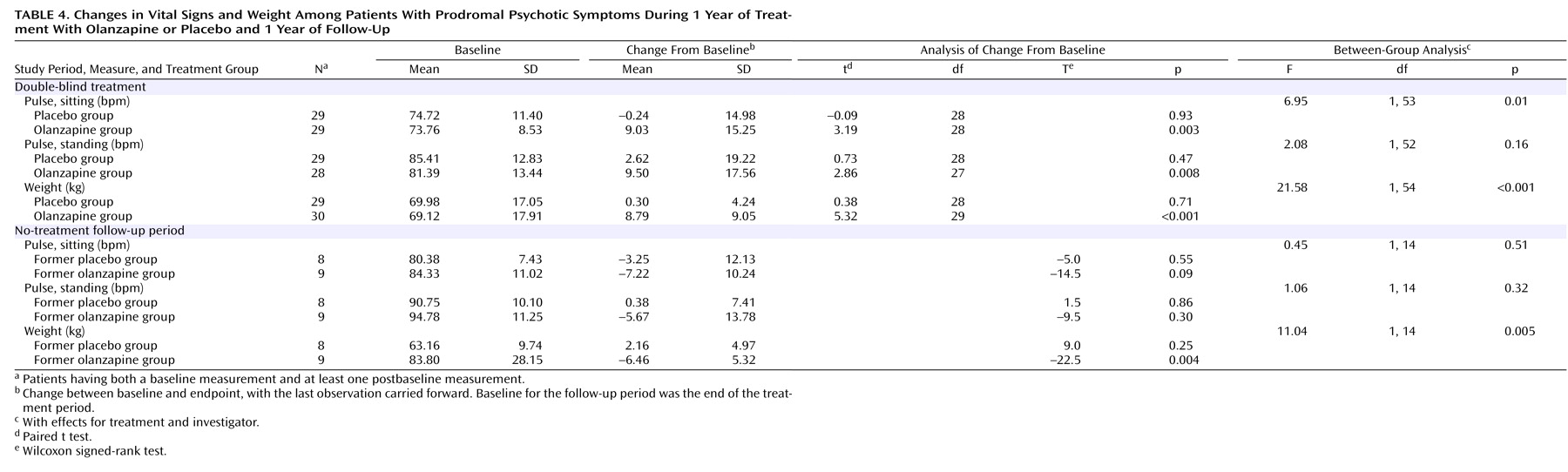
During study period 2, there were no significant differences between treatment groups in mean baseline-to-endpoint changes for extrapyramidal symptoms, in the proportion of patients with treatment-emergent extrapyramidal symptoms, or in the proportion of patients with normal baseline ECG results and abnormal postbaseline results. Of 35 laboratory test results analyzed, there were no values out of the normal range for either group, including values for blood glucose and cholesterol. There were significant differences between groups in mean baseline-to-endpoint changes (with the last observation carried forward) for five measures during study period 2: alkaline phosphatase (F=5.96, df=1, 50, p=0.02), γ-glutamyltransferase (F=6.29, df=1, 50, p=0.02), hematocrit (F=4.50, df=1, 47, p=0.04), inorganic phosphorus (F=5.30, df=1, 48, p=0.03), and uric acid (F=7.96, df=1, 50, p=0.007). There were significant differences between groups in changes on two measures during study period 3: γ-glutamyltransferase (F=6.07, df=1, 14, p=0.03) and lymphocyte count (F=5.95, df=1, 14, p=0.03). However, these laboratory changes were not clinically meaningful. (Laboratory results are available from the first author on request.) During study period 2, the rates of two treatment-emergent adverse events were significantly different in the two treatment groups. Fatigue was reported by 29.0% of the patients in the olanzapine group (nine of 31) and by 3.4% of the patients in the placebo group (one of 29) (p=0.01, Fisher’s exact test). Increased weight was noted by 61.3% of the olanzapine patients (N=19) and 17.2% of the placebo patients (N=5) (p=0.001, Fisher’s exact test). During study period 3, there were no significant differences between groups in the treatment-emergent adverse events that occurred.
Table 4 presents findings on pulse and weight for both study periods. The mean change in weight was significantly greater in the olanzapine group than in the placebo group during study period 2 and significantly lower among the former olanzapine group than among the former placebo group during study period 3. During study period 2, the sitting pulse rate increased significantly in the olanzapine patients relative to the rate for the placebo patients; no other significant between-group differences in vital signs were observed.
Number Needed to Treat
The number needed to treat, calculated as the reciprocal of the absolute risk reduction
(24), provides a clinically useful measurement of the effect of preventive treatment by determining the number of patients a clinician must treat to prevent an adverse event (in this case, psychosis). For our purposes, the number needed to treat was defined as the number of prodromal patients who needed to be treated with olanzapine to prevent conversion to psychosis in the double-blind year. For this study, the number needed to treat equaled 4.5.
Discussion
Study Elements
To our knowledge, this is the first double-blind, placebo-controlled trial that tested specific effects of an atypical antipsychotic on patients with prodromal symptoms of schizophrenia. Also, it monitored patients with a scale sensitive to attenuated schizophrenia-spectrum symptoms, thereby testing the efficacy of treatment for prodromal symptom severity.
The patients were largely adolescent, male, functionally compromised, and displaying symptoms similar to those seen in psychosis but less severe and restrained by intact insight. They provided little evidence of affective disturbance, were almost completely free of substance abuse, and came from families that were largely intact, that were often familiar with mental illness, and that were concerned for their struggling relative and motivated to find help. These characteristics aided recruitment but also may have limited the generalizability of the results to other at-risk populations.
Delaying or Preventing Psychosis
The study patients were at high risk, as 26.7% and 35.0% developed psychosis by the end of the treatment year and posttreatment year, respectively. In the treatment year (study period 2), although not significantly different from placebo, olanzapine had a lower rate of conversion to psychosis (olanzapine: 16.1%, placebo: 37.9%) and a lower hazard of converting to psychosis than the placebo group. In study period 3, the conversion rates were not significantly different (33.3% versus 25.0%). In comparison, significantly fewer experimental (10%) than control (36%) patients converted to psychosis in the first 6 months of the trial reported by McGorry et al.
(4) .
Is the transition to psychosis delayed or prevented by treatment? If olanzapine treatment is palliative and simply delays onset (the way antipsychotics delay relapse in established schizophrenia), then removing the drug should result in a substantially increased rate of conversion. If treatment truly prevents psychosis, then removing the drug should not be followed by many (if any) conversions to psychosis. Our study proved to be underpowered to answer this question. In the no-treatment second year, only 17 patients remained in the trial. Five patients converted to psychosis in the second year—three former olanzapine patients (33.3%) and two former placebo patients (25.0%). These numbers and rates are too small for meaningful comparisons.
Two findings, nevertheless, suggest that we are seeing delay, not prevention. First, if exposure to the drug is truly preventive, we would expect to find few, if any, former olanzapine patients converting in study period 3. In fact, three of nine patients (33.3%) did convert. Second, the prodromal symptoms in the former olanzapine group returned to a significantly higher level of severity when the drug was stopped. Exacerbation of prodromal symptoms would not be expected if time-limited pre-onset treatment was truly preventive.
We were surprised at how operationally benign the process of conversion was. In all cases, patients with conversion to psychosis accepted our judgment that open-label drug treatment was necessary. They complied with treatment without requiring hospitalization, missing significant time from work or school, or disrupting their social and familial networks. It may be that these patients were able to make the transition smoothly because they had an established relationship with us and because conversion was declared at a point where loss of insight was recent and irrational behaviors were still containable.
Reducing Prodromal Symptom Severity
Within their treatment group, the patients receiving olanzapine had significant improvements during study period 2 in the severity of positive and total prodromal symptoms, a finding not seen within the placebo group. Significant differences in positive prodromal symptoms between groups (in favor of olanzapine) were also seen from weeks 8 to 28 of the treatment year. It is possible that after week 28 the remaining study groups contained higher fractions of patients with false positive prodromes.
Our results cannot be compared directly with those of McGorry et al.
(4) because their design did not allow for evaluation of treatment efficacy for symptoms.
Adverse Events
The most significant treatment-emergent adverse event was weight gain. This replicates similar findings in patients suffering from schizophrenia. The weight gain in our study, however, was greater than in a previous study
(25) and may relate to the low age of the subjects
(26) . As in other studies of olanzapine, the rates of treatment-emergent extrapyramidal symptoms were not significantly different between drug and placebo
(27) .
Caveats and Implications for Research in the Prodrome
The limitations of this clinical trial include the lack of a structured axis I assessment at baseline and follow-up and the interrater reliability testing of the Structured Interview for Prodromal Syndromes at only one site. Of importance is that the study addressed an essentially new clinical entity, which required designing new “prodromal” assessment instruments and a new definition of psychosis onset. Other limitations include the fact that our subjects were recruited from families with high rates of mental illness, restricting the ability to generalize the findings to other populations. It is also possible that weight gain may have revealed treatment assignment even though the raters were blinded.
Clear and persistent problems emerged regarding recruitment, group size, and power. Recruitment was difficult because a true positive prodrome is essentially an earlier form of first-episode schizophrenia, the incidence of which is uncommon. Furthermore, the public and treating clinicians are largely unaware of this syndrome’s clinical manifestations and referral possibilities. Compounding the recruitment challenge is the variable fraction of patients with true versus false positive prodromes in each “prodromal positive” study group. Studies of the natural history of such subjects show different rates of conversion and, therefore, the likely ratio of true to false positive at-risk patients in each group. In such studies, conversion rates have varied from 20% to 54% over periods of up to 2 years
(3,
4,
8,
9,
12) .
We originally estimated an overall first-year rate of conversion to psychosis of 30% and hypothesized that twice as many placebo-treated patients as olanzapine-treated patients would convert. Power analysis with such parameters suggested 180 patients (80% power) or 80 patients (50% power). After 3.5 years of recruitment at four sites, we stopped at 60 patients, which corresponded to 39% power for testing treatment effects on transition to psychosis and on prodromal symptom severity. It also meant insufficient power to test whether the treatment was palliative versus preventive. Furthermore, because of the high dropout rate during the trial and the fact that the conversions to psychosis did not occur at the same time points, the a priori plan to use a Fisher exact test to assess the treatment difference in the rates of conversion to psychosis was not appropriate for the final data. Therefore, the results from the treatment comparisons based on the Fisher exact test should be interpreted with caution and should be supplemented with the results from the post hoc analysis with the Cox proportional hazards model.
It is striking that all of the olanzapine patients whose symptoms converted to psychosis did so within the first 4 weeks of the clinical trial. These patients were among the most symptomatic, suggesting several possibilities. First, some of these patients may have been misclassified as prodromal when they were psychotic, especially because patients may be reluctant to discuss psychotic symptoms on initial evaluation. It is possible that some patients were already psychotic but unable to communicate this until, paradoxically, they received sufficient olanzapine to convey effectively their state of mind. Finally, some of these patients may have been on the cusp of psychosis and were not medicated rapidly or sufficiently enough to forestall conversion.
Further studies are clearly needed to replicate and extend our findings, which suggest strategies for future research of this type. First, ongoing compliance should be monitored carefully and should include blood tests to determine the drug levels in converting patients at the time of conversion. Second, it may be prudent to design 2- to 4-week placebo or no-treatment lead-in periods before randomization, excluding patients who meet the criteria for being psychotic within this period. Third, future studies should use larger study groups and longer assessment times to determine whether a medication could prevent psychosis onset if continued for longer than 1 year. Fourth, power should be enhanced by 1) determination of further markers of risk for psychosis to improve the identification of the truly at-risk subset of any clinically “prodromal” subject group or 2) collaboration among multiple sites and pooling of subjects
(28) .
Conclusions
Although the primary objective of demonstrating a significant treatment difference in the rate of conversion to psychosis was not met, the nearly significant difference between treatment groups in the conversion rate points to the possibility that olanzapine might reduce the rate of conversion to psychosis and delay the onset of psychosis. For estimating benefit, the number needed to treat of 4.5 suggests that between four and five persons meeting the criteria for prodromal symptoms need to be treated in order to prevent one conversion to psychosis over the 1 year of treatment. This is comparable in magnitude to the number needed to treat reported in the clinical trial of risperidone and cognitive behavior intervention by McGorry et al.
(4) . For estimating risk, the treated patients will gain an average of 13% in body weight in the first year, but they will not display blood laboratory values suggestive of risk for diabetes or cardiovascular disease. Being heavier, these young people may be at higher long-term risk for the metabolic syndrome, but they do not display this during the first year of treatment. Ratios of longer-term benefit and risk require more clinical trials with variable durations of clinical exposure.
Overall, we feel that this clinical trial demonstrates that the benefits of pre-onset identification and treatment outweigh the risks to a degree sufficient to endorse future clinical trials (in academic centers with programs for early detection and intervention in psychosis) so that definitive recommendations on use of antipsychotics to treat the prodromal phase of schizophrenia can be made.