Autism is a neurodevelopmental disorder that has a strong, complex genetic component. Estimates suggest that multiple genes contribute to risk, but to our knowledge, no single gene of significant effect has yet been identified. Several genomic screens identified the 2q31–33 region (170–198 cM) as a consensus linkage region
(1 –
4) .
A number of potential candidate genes map within this region, and several have been evaluated for association with autism
(3 –
5) . These include
SLC25A12 (approximately 172 megabases [Mb]), which encodes a mitochondrial aspartate/glutamate carrier expressed in the brain. Ramoz et al.
(5) reported significant association between autism and two single nucleotide polymorphisms (SNPs), rs2056202 in intron 16 (3′ end of the gene) and rs2292813 in intron 3 of
SLC25A12, as well as the haplotype for the two SNPs (p=0.003, p=0.05, and p=0.002, respectively). To investigate these findings in our independent data set, we analyzed these two SNPs and seven other SNPs within this gene, covering the entire transcriptional unit.
Method
Association analyses were performed on 327 Caucasian families with autistic offspring, including trios (one affected child and two parents) and multiplex families (two or more affected children and parents). A complete description of the study was provided to the subjects, and written informed consent was obtained. The families included 230 from Duke University and 97 from Vanderbilt University. All of the affected offspring met the DSM-IV diagnostic criteria for autism. Detailed diagnostic evaluations of the family data have been previously described
(5) . Approval was obtained from both the Duke University and Vanderbilt University Medical Center institutional review boards.
Blood was obtained from the patients and other family members, and DNA was extracted from whole blood by using standard protocols. Nine SNPs (rs2292813, rs970948, rs1878583, rs11757, rs6758704, rs6433317, rs3821095, rs2056202, and rs925881) within SLC25A12 were selected from the SNP database of the National Center for Biotechnology Information (www.ncbi.nlm.nih.gov/SNP/). All SNPs were ordered as TaqMan (Applied Biosystems, Foster City, Calif.) assays and genotyped according to the manufacturer’s recommendation.
Hardy-Weinberg equilibrium was assessed, and pairwise linkage disequilibrium (D′ and r
2 ) between markers was calculated. The pedigree disequilibrium test and the genotype pedigree disequilibrium test were used to examine disease association. The family-based association test
(6) was used for haplotype analyses. To run the haplotype analyses, we obtained global scores for a pairwise haplotype analysis that encompassed all SNPs within the gene. For each pair of SNPs, haplotypes observed in fewer than 10 families were not considered
(6) . TDT-GENEHUNTER (GENEHUNTER version 2.1; http://www.fhcrc.org/science/labs/kruglyak/Downloads/) was also used as a measure for linkage.
Results
Tests for Hardy-Weinberg equilibrium deviations were calculated for each marker in two groups of affected and unaffected individuals selected at random from each family. No polymorphism showed evidence of deviation from Hardy-Weinberg equilibrium in any data set.
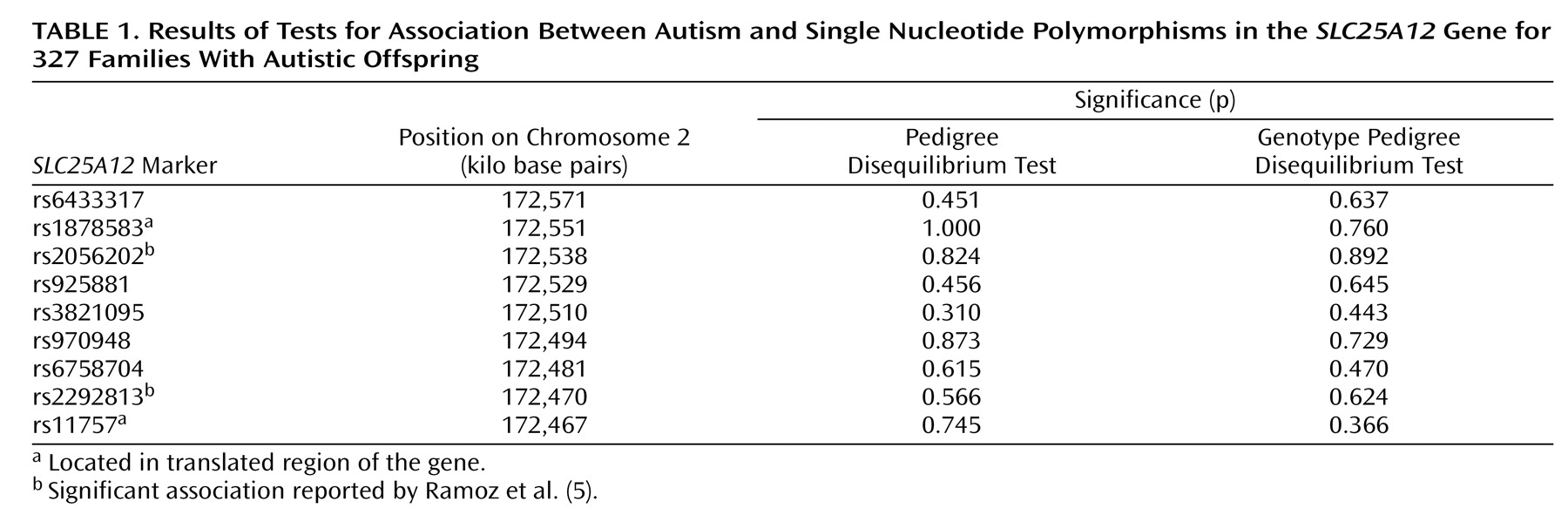
The pedigree disequilibrium test and the genotype pedigree disequilibrium test showed no association with autism for any of the SNPs within
SLC25A12, including the two SNPs for which Ramoz et al. found significant association (
Table 1 ). In addition, there was no evidence of association (p≤0.05) with any specific pairwise haplotype according to haplotype analyses with the family-based association test.
To ensure that this lack of association was not due to a lack of informativity of the SNPs in our data set, the number of families informative for each SNP was calculated (a family was considered informative if it had at least one heterozygous parent). The number of informative families ranged between 124 (rs2292813 and rs1878583) and 249 (rs3821095). To capture linkage information, we analyzed the families that showed positive lod scores (logarithms of the odds ratios for linkage)
(4) for the significant SNPs in the study by Ramoz et al.
(5), rs2292813 and rs2056202. Analysis within these “linked” families also failed to detect significant association (pedigree disequilibrium test: p=0.09 and p=0.91 for 18 and 21 informative families, respectively). Additionally, a transmission disequilibrium test was run to assess linkage in the entire data set. The results were nonsignificant. A high amount of linkage disequilibrium (D′≥0.93, r
2 ≥0.76) was found across the tested SNPs.
Discussion
We analyzed a panel of nine SNPs in
SLC25A12, spanning the 110-kb transcriptional unit, in 327 autism families independent of those previously reported by Ramoz et al.
(5) . Markers were selected to span the gene with an average spacing of 10 kb. The selected markers included a silent mutation in the 5′ untranslated region (rs1878583) and an SNP in the 3′ untranslated region (rs11757), in addition to rs2056202 and rs2292813
(5) . We found no evidence of association for any of the markers analyzed or for any specific pairwise haplotype in these data.
Ramoz et al. also reported positive linkage to the region and to the two significant SNPs in
SLC25A12 (approximately 172 Mb). We previously reported evidence for linkage to this same region within a subset of our families
(4), with a peak lod score at D2S1776 (approximately 169 Mb), but in the current investigation we did not find evidence of linkage for any of the
SLC25A12 SNPs within the overall data set in this study. Moreover, we did not find evidence for significant association in the families with positive lod scores for
SLC25A12 SNPs.
There are several possibilities to consider when interpreting the lack of consistency between these two studies. The causative variation in SLC25A12 could be an SNP different from the SNPs analyzed by Ramoz et al. but in strong linkage disequilibrium with them. However, given that we analyzed SNPs covering the entire transcriptional unit, we would still expect to see an association. In addition, the association described by Ramoz et al. could be caused not by variation in SLC25A12 but, instead, by variation in a nearby gene, which is in linkage disequilibrium with these two SNPs in their study group. It is also possible that the findings of Ramoz et al. could be due to a type I error, but this seems unlikely given the significance of their results.
Population differences could account for the different results. Owing to chance sampling, the Ramoz et al.
(5) data set may include a subset of families that show the effect, or a real but unknown difference may exist between our two study groups. Undetected phenotypic differences between the two groups of families could exist, as it is well established that clinical symptoms vary widely within this diagnosis. Subtle differences in the expression of the disease in the various subgroups in the two studies could have significant genetic implications. Further investigation into the phenotype of families contributing to the association is needed in order to clarify these issues. In summary, we have failed to observe association findings similar to those reported by Ramoz et al.
(5), but we cannot rule out the possibility that
SLC25A12 has a modest effect in a subset of families.