Psychological Versus Biological Clinical Interpretation: A Patient With Prion Disease
Case Presentation
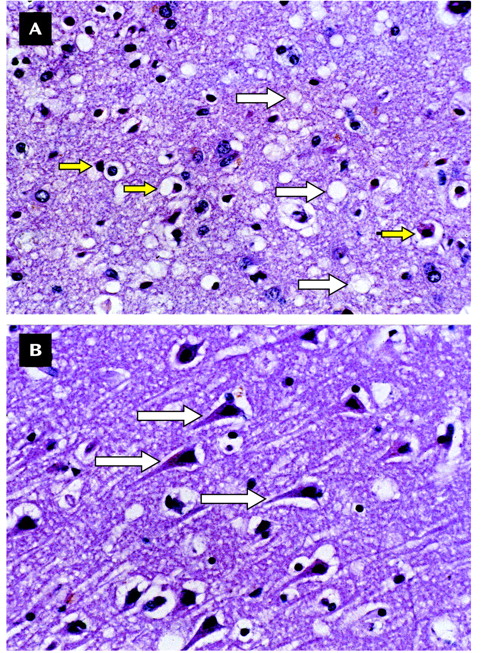
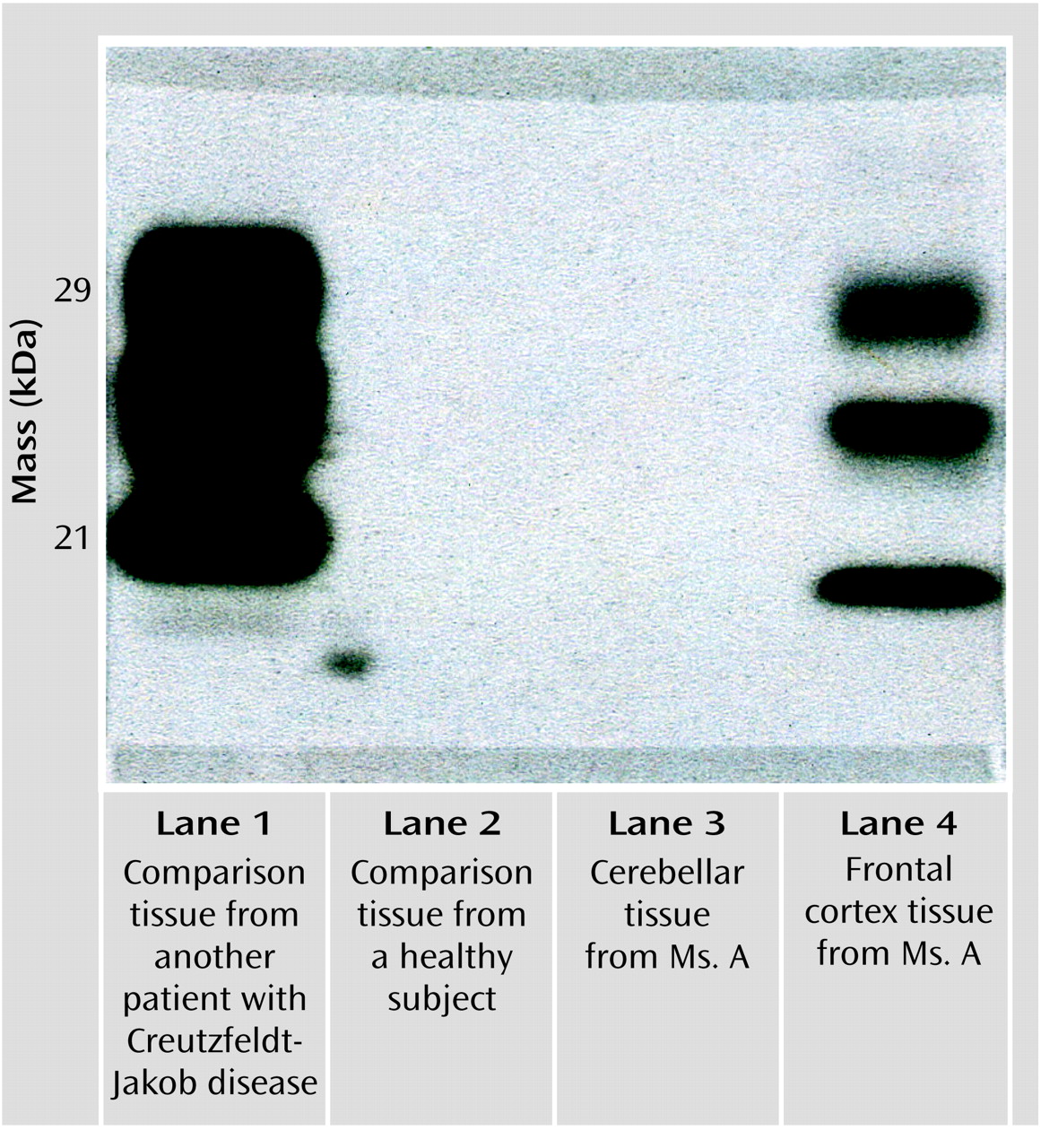
Ms. A was a 49-year-old Caucasian woman who had never married and who had been a communications product engineer. Before onset of her symptoms, she had never seen a psychiatrist, never been diagnosed with a psychiatric disorder, and never been treated with a psychotropic medication. Her medical history was notable only for a febrile illness that may have been an encephalitis of uncertain etiology after a trip to South America when she was in her 20s. There was no family history of psychiatric illness, epilepsy, or neurological or neurodegenerative disorders.Ms. A had grown up as the youngest of three children. She described her relationship with her two brothers as an adult as “distant.” Her relationship with her parents was reported to have been “good,” although additional details were lacking, and both parents were deceased. She had no history of physical or sexual abuse, but she felt that she had been excessively teased by her brothers while growing up, which left residual resentment even late into her adult life. She did not date frequently in either her teens or 20s. She had a number of heterosexual romantic relationships but described these as superficial and brief. She noted that she had always been sexually interested in women, but until just before the onset of the symptoms leading to hospitalization, she had never had a same-sex romantic relationship. The revelation of her homosexuality to her family was poorly received. She described it as “cold,” and she felt criticized and ashamed.Ms. A had begun her employment with a communications company as a telephone operator and had advanced over the next 20 years until she became a high-level manager in research and development. Ultimately, she was recruited into an upper-level management position with a competitor in the field. She traveled frequently to Europe as part of her new job. During this transition in her career, she met a woman with whom she hoped to have a fulfilling romantic relationship and thus estranged herself from her family.In the context of these psychosocial stressors and after a flight from Europe, she developed an acute onset of lower-back pain while lifting her laptop computer out of an overhead bin. Soon after she saw a chiropractor for lower-back pain; upon examination, the practitioner noted some weakness of the lower extremities. She received no benefit from the chiropractic visits and began to note the onset of new symptoms. These included a tremor “all over [her] body” and weakness in “big muscle groups.” This made ambulation difficult and was the beginning of a slow deterioration in gait. She made an appointment with her primary care physician. A workup included magnetic resonance imaging (MRI) of her spine, which showed a mild lumbar right-paramedian bulge that was judged to be inadequate to explain her symptoms. Her laboratory tests included a CBC, a full metabolic panel, including liver function tests—all of which were found to be within normal ranges—a measurement of erythrocyte sedimentation rate (4), and a rheumatoid factor check (negative result). These results were obtained almost 2 months after Ms. A’s original lower-back strain.Ms. A’s medical and emotional needs became overly burdensome to her partner, which led to the end of their romantic relationship. Ms. A continued to experience weakness in her lower extremities and ambulation difficulties. Her physician ordered an MRI of the brain, which was normal except for evidence of chronic sinusitis in the left maxillary sinus. An MRI of her cervical spine was also performed, and the results were found to be normal. Additionally, a lumbar puncture was performed as the diagnostic considerations expanded to include multiple sclerosis, encephalitis, neurosyphilis, subacute sclerosing panencephalitis, and acute idiopathic polyneuritis. Results of tests regarding CSF glucose, cell cytology, protein, gram stain, immunoglobulin M, immunoglobulin G, fluorescent treponemal antibody absorption, and immunoglobin banding were all negative.Despite reassurance that these symptoms would likely remit, Ms. A continued to experience more weakness and ataxia. She was seen by another internist and diagnosed with atypical multiple sclerosis. She was given a trial of methylprednisolone sodium succinate and noted a brief period of symptom improvement. She was seen by a neurologist 1 month later, and an EEG was ordered. Again, the result was unrevealing.Five months after her initial symptoms, Ms. A complained of diplopia, difficulty swallowing, and frequency of urination in addition to her symptoms of leg weakness, tremor, and gait difficulties. She saw another neurologist whose differential diagnosis included Creutzfeldt-Jacob disease. She was then seen by an associate of a national authority on prion disease, who felt that Creutzfeldt-Jakob disease was a “low-probability” diagnosis, but a measurement of blood mercury level was recommended, which was also found to be negative.At this point Ms. A’s treating physician began to seriously consider that these symptoms might have a psychological component, which led to a psychiatric evaluation. About 6 months from the onset of her symptoms, Ms. A completed a neurocognitive and psychological battery of tests. The results were as follows: score of 30/30 on the Mini-Mental State Examination (MMSE), a score of 33/33 on the Blessed Dementia Scale (5), and a Sensory Perceptual Screening (6) examination performed without errors. She completed the Wechsler Memory Scale—Revised (7). Her score fell within the average to high-average range, and she was judged to have high-average intellectual abilities. Also of note was that her receptive and expressive language abilities were found to be intact, her conversational speech was fluid and nonphasic, her digit repetition was high average, her oral calculations were average, and her attention and concentration were normal. The only abnormal finding in this test battery was on the MMPI-2, which showed elevated depression.Although Ms. A complained of weakness and an unstable gait, results of formal neurologic examinations continued to be completely normal. In addition, Ms. A’s complaints of waxing and waning strength and difficulty walking were difficult to explain. At this time, she was given axis I diagnoses of major depression and conversion disorder. Ms. A was given fluoxetine for depression and buspirone for anxiety. She was thereafter discharged to a residential psychiatric facility.Nearly 8 months after the first appearance of lower-back strain, Ms. A was seen by a specialist in dissociative disorders and psychosomatic illness. He hypnotized Ms. A and noted that she was highly hypnotizable and that, under hypnosis, her symptoms improved. She continued to decompensate and was transferred from the residential facility to a skilled nursing facility secondary to functional deterioration and an inability to complete activities of daily living without assistance. This deterioration continued until Ms. A was again hospitalized, this time on the Behavioral Medicine Unit at Stanford University Hospital. At that time, she was confined to a wheelchair. She complained of waxing and waning dysarthria and difficulty swallowing, an ataxic-like gait, and tremors that would move over her entire body, which she would refer to as “convulsions.” She also had occasional complaints of diplopia, for which she would compensate by closing one eye, and frequent squinting. She continued to experience subjective weakness in her lower extremities, which varied in degree, although it was always present.At this time, both Ms. A and her family agreed that her mood was normal. Her score on the MMSE at admission was 24/30; she appeared to be poorly motivated to complete the test. Her insight was judged to be good, and she was reported to say that she was motivated to determine the cause of her disability so she could return to work. She admitted that being ill had brought her family back to her and that her ex-lover was again in frequent contact, both of which pleased her deeply. Upon examination her tremors were noted to wax and wane depending on the content of her conversation. Her diagnoses at admission were conversion disorder and major depression, single episode, moderate, nonpsychotic, and in remission with a regimen of fluoxetine and buspirone. Psychological testing was ordered, as was a follow-up neurology consultation.Upon completion of neurological and psychometric evaluations, Ms. A’s primary diagnosis remained conversion disorder. Supporting this diagnosis were fluctuating and at times bizarre symptoms: waxing and waning memory deficits, an inability to ambulate without assistance despite adequate strength, back pain, eye squinting, and tremor, but no evidence of spasticity or cerebellar abnormalities. There was also some concern over factitious elements, such as a new complaint of life-long auditory and visual hallucinations.Ultimately, it was decided to proceed with a behaviorally oriented rehabilitation program. This involved aggressive physical therapy, occupational therapy, and participation in groups and activities, with the focus on treating Ms. A’s functional disability. Furthermore, with the intent of both confirming and treating the conversion disorder, it was explained to Ms. A that in the setting of such an intensive rehabilitation program, her functional ability should improve if it was simply related to an underlying medical etiology, such as the apparent severe muscle deconditioning she had experienced. It was also explained to Ms. A that if she did not improve after undergoing such an intensive rehabilitation program, it would be owing to either a lack of participation or motivation on her part or because her condition was purely the result of a psychological disturbance. That is to say, improvement would confirm a medical etiology. This behavioral approach to treating conversion disorder was based on a review of the literature and the apparent success of the use of this “double-bind” model (3).The neurology service was consulted soon after Ms. A was hospitalized. They found her to have normal conjugate eye movements without nystagmus, although she complained of diplopia. Her palate lifted symmetrically, her gag reflex was intact, and a fluctuating dysarthria in her speech was described as “clear” at times. She was noted to have decreased muscle bulk on the anterior tibialis muscles bilaterally and a slight contracture of the left Achilles tendon, both attributed to deconditioning. Her muscle strength was intact, with bilaterally symmetric deep tendon reflexes. She had intermittent tremors of her trunk and extremities, both at rest and with movement, without an apparent pattern. She had no cerebellar abnormalities; e.g., heel-to-shin and finger-to-nose movements were intact. While standing Ms. A was unable to place her left heel to the floor, was unsteady, and was unable to stand unassisted. She appeared to be cognitively intact, although her affect ranged from tearful to “flattened.” Noting her prior workup and neurologic evaluations, the neurology service assessed the observed tremors and other symptoms as highly elaborated and “functional” in nature. There was noted a lack of cooperation with the funduscopic examination, which was complicated by Ms. A’s frequent blinking. The neurology service emphasized that at that time there was no evidence of nervous system disease, concurred with a diagnosis of conversion disorder, and recommended continued behaviorally based treatments.With great encouragement, Ms. A was at times able to ambulate somewhat better and at other times was only able to ambulate with two-person support. Her speech would fluctuate in terms of intelligibility, her tremors would wax and wane, and her ability to perform on the MMSE would change as well. Some of the variability was clearly associated with anxiety about issues and concerns expressed in the content of her speech. She clearly responded to decreased nursing attention with vocalizations but not always with words, sometimes moaning plaintively. While eating she was observed to throw her head back, causing her to choke on her food; however, with consistent reinforcement, she would keep her chin down while chewing and have no difficulty. Because of concerns over her apparent swallowing difficulties, a videofluoroscopic study was performed and an occupational therapy swallowing examination completed. Both showed normal swallowing reflexes but noted that she would tilt her head back and choke at times. This was considered to be volitional behavior.There were also attempts at managing her condition with psychotropic medication. Ms. A had had a good response to fluoxetine for her depression, and this treatment was continued and the dose increased. The buspirone therapy was continued, and olanzapine was added at a dose up to 10 mg, given at night, mostly for episodes of agitation and concern that Ms. A was at times psychotic. None of these medication changes appeared to change her course of illness.Approximately 6 weeks after Ms. A’s admission, the psychiatry service noted waxing and waning primitive reflexes. In addition, she began to exhibit progressively more disinhibited behavior, such as throwing food and smearing it on her face, along with choking sounds. She developed an exaggerated startle response to innocuous stimuli.Another MRI and EEG were performed, and again the neurology service was consulted. Ms. A had an essentially normal examination. The service noted slurred speech but felt it was functional and that no evidence of dysarthria was present. Ms. A knew the date but appeared to be disoriented to place. The neurology service did not note any disconjugate gaze but raised questions about subtle right nasolabial fold flattening, oral dyskinesias, and uncoordinated tongue movements. There also was a concern over a subtle right lower-extremity rigidity that had not been noted previously. Additionally, mild, right-greater-than-left intention tremor was noted.Ms. A’s repeat MRI of the head resulted in a normal scan without evidence of ventricular enlargement or expanded sulci. The neurology team noted that the buspirone may have caused her tremor, referring to it as tardive dyskinesia, and thought both fluoxetine and olanzapine may have contributed to it as well. They recommended simplification of the medication regimen.Earlier in her hospitalization, Ms. A had been administered an MMPI-2. The results of the entire neuropsychological battery were now considered to be invalid because of her excessive endorsement of symptoms. In addition, a Rorschach test given Ms. A was deemed unscorable. She tended to tell long, convoluted stories about the inkblots and could not be redirected. She used what appeared to be neologisms (e.g., “torcle bug” and “buldar”), but when she was questioned, she would laugh and say that she had made them up by combining words. Ms. A was dysarthric and difficult to understand. She tended to avoid responding to questions with direct answers and appeared to be somewhat disinhibited; e.g., she asked the female examiner out on a date.Given the lack of information gained from administration of these tests, the examiner requested copies of the raw data from Ms. A’s previous neuropsychological testing. Her MMPI-2 results, taken 4 months earlier, were notable only for depression and lacked the expected elevation in somatic preoccupation, denial, repression, and endorsement of neurological symptoms that is often found in individuals with conversion disorder.Subsequently, psychological testing was readministered, and this time it was considered to be valid. Ms. A was given the shorter Millon Clinical Multiaxial Inventory—III (8), the Rorschach test, and the Thematic Apperception Test. The results of the Millon Clinical Multiaxial Inventory—III were considered to be valid, but Ms. A appeared to answer it with a bias toward magnifying her symptoms, as it indicated the presence of a significant mental disorder. Her profile suggested a personality disorder with narcissistic, avoidant, and paranoid features. Her Rorschach test results had 19 scorable responses and, this time, was notable for linear responses and an absence of neologisms. Ms. A exhibited considerable effort and demonstrated complexity in her thought process, as well as an ability to synthesize information. She had some difficulty with perceptual accuracy and demonstrated a tendency to become easily disorganized.Ms. A continued to fare poorly, her grooming was very poor, and her ability to perform simple activities of daily living was severely impaired. Despite aggressive behavioral strategies to treat a presumptive conversion disorder, no progress was seen. Repeated neurologic consultations were inconclusive. An MRI and EEG showed no abnormality. After exhaustive review of Ms. A’s care, strategies to improve her function, and attempts to define a possible medical etiology, it was reluctantly agreed that no progress had been made in the prior 6 weeks of intensive therapies. Plans were made to have her moved to a skilled nursing facility.Ms. A’s response to this proposed move was dramatic. There was an increase in nonverbal vocalizations, choking, disinhibited outbursts, difficulty following directions, and tearfulness. She began to look more abulic, with more frequent and severe difficulties with chewing and swallowing. With the expected discharge date looming, another EEG was ordered.It was decided to place a nasogastric tube for feeding. However, less than 24 hours after insertion, it was taken out by Ms. A. Another was placed, with a similar result. Faced with the difficulties of having Ms. A undergo even a minor procedure for the presumed conversion disorder, her unwillingness to allow the nasogastric tube to remain in place, and ambivalence about proceeding to percutaneous endoscopic gastrostomy placement in an individual with a psychosomatic illness, we decided to initiate another trial of oral feedings. That day Ms. A ate both lunch and dinner without significant difficulty.Ms. A was found later that night in cardiopulmonary arrest. It occurred almost 90 minutes after dinner, which had been observed by nursing staff and described as uneventful. After dinner that night, Ms. A had been heard choking; although her oropharynx had been examined and was found to be clear of food and debris. She was found cyanotic approximately 30 minutes later, without respiration or pulse. Suction performed during cardiopulmonary resuscitation revealed small bits of food—apparent evidence of aspiration. Ms. A was presumed to have asphyxiated because of an obstructed airway. It was thought possible at the time that Ms. A may have moved some food into her chair and later put it into her mouth and choked, suffering asphyxiation, cardiopulmonary collapse, and near-death. She was resuscitated and sent to the intensive care unit, where about 36 hours later she was declared brain dead by EEG.Ironically, the EEG ordered earlier in the week was read at the same time as the EEG from the ICU. The former was noted to be abnormal and interpreted as showing excessive theta activity in bilateral temporal areas that was consistent with an encephalopathy. The EEG report specifically mentioned the possibility of a rapid neurodegenerative process in the differential. Ms. A was removed from life support after a lengthy meeting with her family, and she expired shortly thereafter.The family granted permission for an unrestricted autopsy. On general autopsy, Ms. A was found to have acute and organizing bronchopneumonia, with fragments of vegetable material in the bronchus consistent with prior aspiration. The results of gross examination of the external brain and coronal sections was found to be entirely normal, with no evidence of inflammation, neoplasm, atrophy, or white matter changes. However, microscopic examination revealed changes indicative of prion disease; namely, sections of the frontal, hippocampal, and occipital cortices showed moderate neuronal loss, reactive astrocytosis, and variably sized vacuoles within the neuropil, also know as spongiform change (Figure 1A). No kuru-type plaques were seen; no inflammation was observed. The underlying white matter showed patchy myelin loss and spongiform change. In addition, many of the sections revealed evidence of acute ischemic/hypoxic injury, with shrunken neurons displaying hyperchromatic, indistinct nuclei and eosinophilic cytoplasm (Figure 1B). Sections of the midbrain and brainstem also showed spongiform change, as well as neuronal dropout and gliosis in the substantia nigra and olivary nuclei. A section of the cerebellum showed a normal-appearing granular cell layer, loss and acute ischemic/hypoxic injury of the Purkinje neurons, and mild spongiform change in the molecular layer. The underlying cerebellar white matter had mild spongiform change, and the dentate nucleus showed gliosis and neuronal loss.In compliance with an ongoing surveillance program sponsored by the American Association of Neuropathologists and the Centers for Disease Control and Prevention, tissue was sent for immunodiagnostic studies to collaborators at the newly formed National Prion Disease Pathology Surveillance Center in Cleveland. Western immunoblot analysis revealed the presence of the pathogenic, protease-resistant isoform of prion protein, which confirmed the diagnosis of prion disease (Figure 2). Of interest, this sensitive detection method (9) discerned the protease-resistant isoform of prion protein, as evidenced by the unique three-band pattern revealed after digestion with proteinase K in the comparison tissue from another patient with Creutzfeldt-Jakob disease (lane 1) and in the frontal cortex from Ms. A (lane 4). It found no protease-resistant isoform of prion protein in the cerebellar tissue (lane 3). This might suggest a sampling or technical error or that levels of the prion protein in the cerebellum were possibly too low for detection.
Discussion
Conclusions
Footnote
References
Information & Authors
Information
Published In


History
Authors
Metrics & Citations
Metrics
Citations
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
For more information or tips please see 'Downloading to a citation manager' in the Help menu.
View Options
View options
PDF/EPUB
View PDF/EPUBGet Access
Login options
Already a subscriber? Access your subscription through your login credentials or your institution for full access to this article.
Personal login Institutional Login Open Athens loginNot a subscriber?
PsychiatryOnline subscription options offer access to the DSM-5-TR® library, books, journals, CME, and patient resources. This all-in-one virtual library provides psychiatrists and mental health professionals with key resources for diagnosis, treatment, research, and professional development.
Need more help? PsychiatryOnline Customer Service may be reached by emailing [email protected] or by calling 800-368-5777 (in the U.S.) or 703-907-7322 (outside the U.S.).