Several neurochemical hypotheses have been proposed to account for the origin and symptoms of schizophrenia, including abnormal dopaminergic, γ-aminobutyric acid (GABA)-ergic, and glutamatergic neurotransmission
(1–
5). Evidence supporting an association between glutamatergic dysfunction and schizophrenia has come from pharmacological studies showing that
N-methyl-
d-aspartate (NMDA) receptor antagonists, such as phencyclidine and ketamine, can induce many of the psychotic signs and symptoms of schizophrenia in normal comparison subjects or exacerbate them in patients with schizophrenia
(6–
8). Reports of abnormal glutamatergic neurotransmission in the hippocampus, entorhinal cingulate, and prefrontal cortices
(2,
9–13) of schizophrenic patients and the involvement of the glutamatergic system in learning, memory, emotion, and behavior
(14) have given further credence to the hypothesis of the dysregulated glutamatergic state in schizophrenia.
Similarly, evidence for abnormal GABA-ergic neurotransmission in schizophrenia includes lower GABA uptake and release in the frontal cortex
(15), lower glutamic acid decarboxylase activities and mRNA in some brain regions and neurons
(16–
18), greater [
3H]-muscimol and GABA
A receptor binding
(19,
20), fewer small, putatively GABA-ergic neurons in the hippocampus and the anterior cingulate cortex
(21–
23), and less GABA-transporter-1 protein in axon terminals of chandelier neurons
(24). However, evidence suggestive of greater laminar GABA-ergic activity has also been reported
(25,
26). Thus, in addition to glutamatergic dysfunction, there are concurrent defects in GABA-ergic neurotransmission
(20,
26,
27).
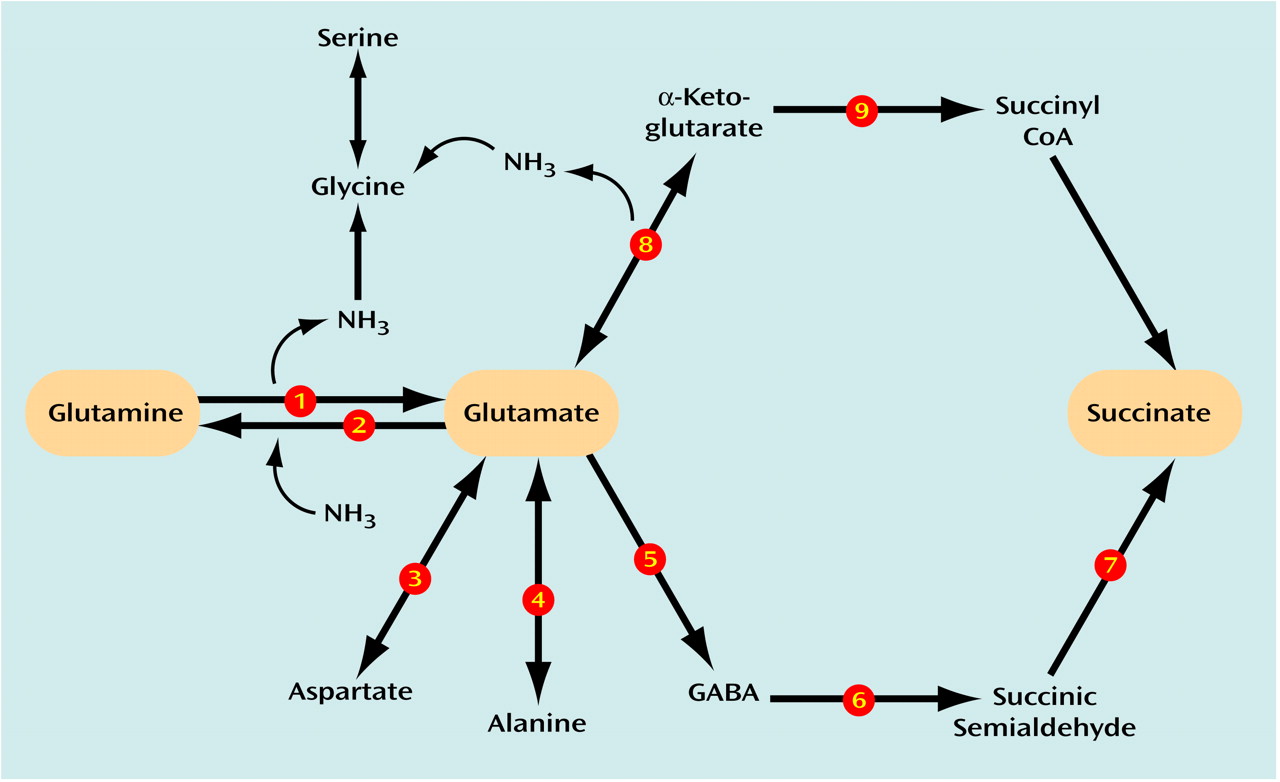
CNS glutamate and GABA metabolism entails a series of integrated synthetic and degradative pathways (
Figure 1). Glutamate, in addition to its role in brain ammonia clearance, serves as an important energy source after its deamination by glutamate dehydrogenase and oxidative decarboxylation by α-ketoglutarate dehydrogenase. Furthermore, glutamate is integrally related to the synthesis of glutamine (by means of glutamine synthetase) and aspartate and alanine (through aspartate aminotransaminase [AST] and alanine aminotransaminase [ALT], respectively). Glutamate can be synthesized by glutamate dehydrogenase, AST, ALT, GABA-transaminase, and phosphate-activated glutaminase, a neuronal enzyme most directly involved in the generation of glutamate pools for neurotransmission. Glutamate is converted to GABA by glutamic acid decarboxylase. Finally, GABA is metabolized to succinate by the combined reactions of GABA-transaminase and succinic semialdehyde dehydrogenase.
Since GABA derives from glutamate and glutamate can derive from GABA, alterations in glutamate metabolism can effect GABA metabolism or vice versa. The overall aim of this study was to confirm that GABA and glutamate metabolism was abnormal in schizophrenic patients and to determine how these activities may be associated with one another. A panel of nine principal enzymes involved in glutamate and GABA metabolism (glutamate dehydrogenase, glutamine synthetase, phosphate-activated glutaminase, α-ketoglutarate dehydrogenase, AST, ALT, glutamic acid decarboxylase, GABA-transaminase, and succinic semialdehyde dehydrogenase) (
Figure 1) was studied concurrently in the same brain region (dorsolateral prefrontal cortex) in the same group of aged antemortem-assessed and diagnosed schizophrenia patients dying of natural causes and normal age-matched nonpsychiatric comparison subjects. The dorsolateral prefrontal cortex
(28) was targeted for study because it has been implicated in neuroimaging studies
(29), it shows significant shrinkage and greater packing density in schizophrenia patients
(30), it has an important role in the mediation of working memory
(29,
31), it is affected by long-term treatment with phencyclidine
(32), and it is a site of interaction between glutamatergic receptors and the mesocortical dopamine system
(33). To test the regional specificity of findings, specimens dissected from the occipital cortex (Brodmann’s area 17) were also assessed in the same subjects.
Results
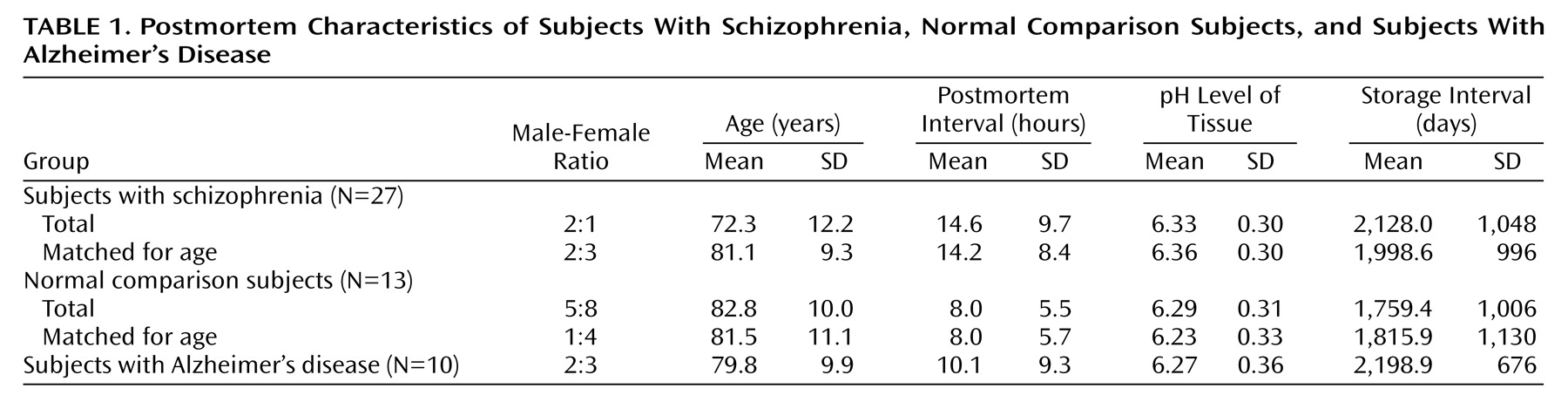
The schizophrenic subjects were younger as a group than the other groups (F=4.03, df=2, 47, p<0.03; Newman-Keuls test, p=0.02) (
Table 1). Between-group differences in postmortem interval, tissue pH, and storage interval were not statistically significant (all p>0.10). Two approaches were taken to control for the group differences in age. First, in statistical analyses that involved comparisons of entire groups, age was entered as a covariate. Second, when statistically significant differences were observed between groups, t tests were repeated on subgroups of 10 normal comparison subjects and 10 schizophrenic subjects that were matched closely for age (
Table 1).
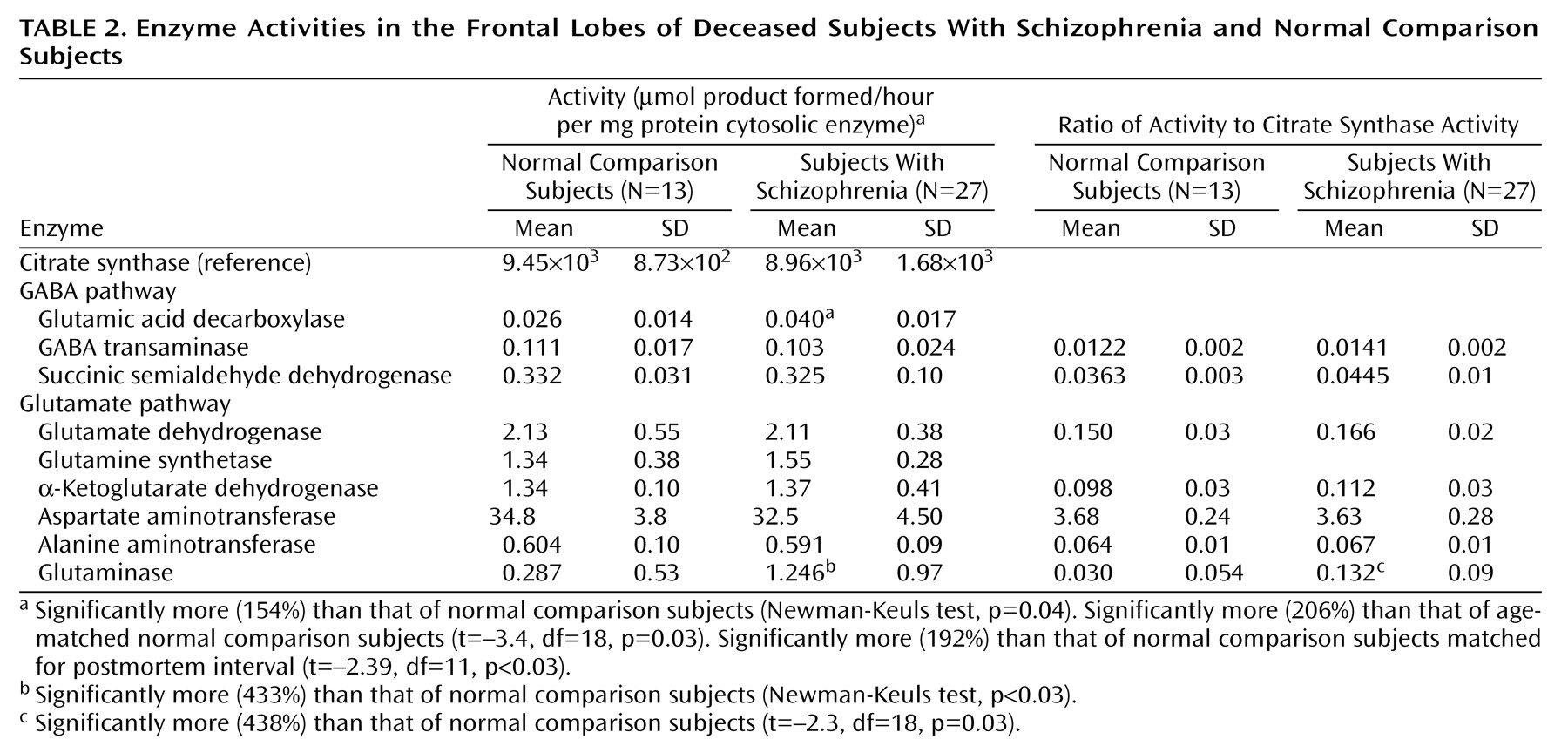
Citrate synthase activities in the dorsolateral prefrontal cortex were calculated per mg of protein and subsequently expressed as units of activity (μmoles product formed/hour per mg of protein) for comparison with other enzymes (
Table 2). No significant differences in citrate synthase activity were observed among the three groups (F=0.96, df=2, 47, p=0.39). Furthermore, there were no correlations between citrate synthase activity and postmortem interval, tissue pH, or storage time (r<0.24, N=40, p>0.12). Results of assays for mitochondrial enzymes were expressed in relation to both citrate synthase activity and to protein content.
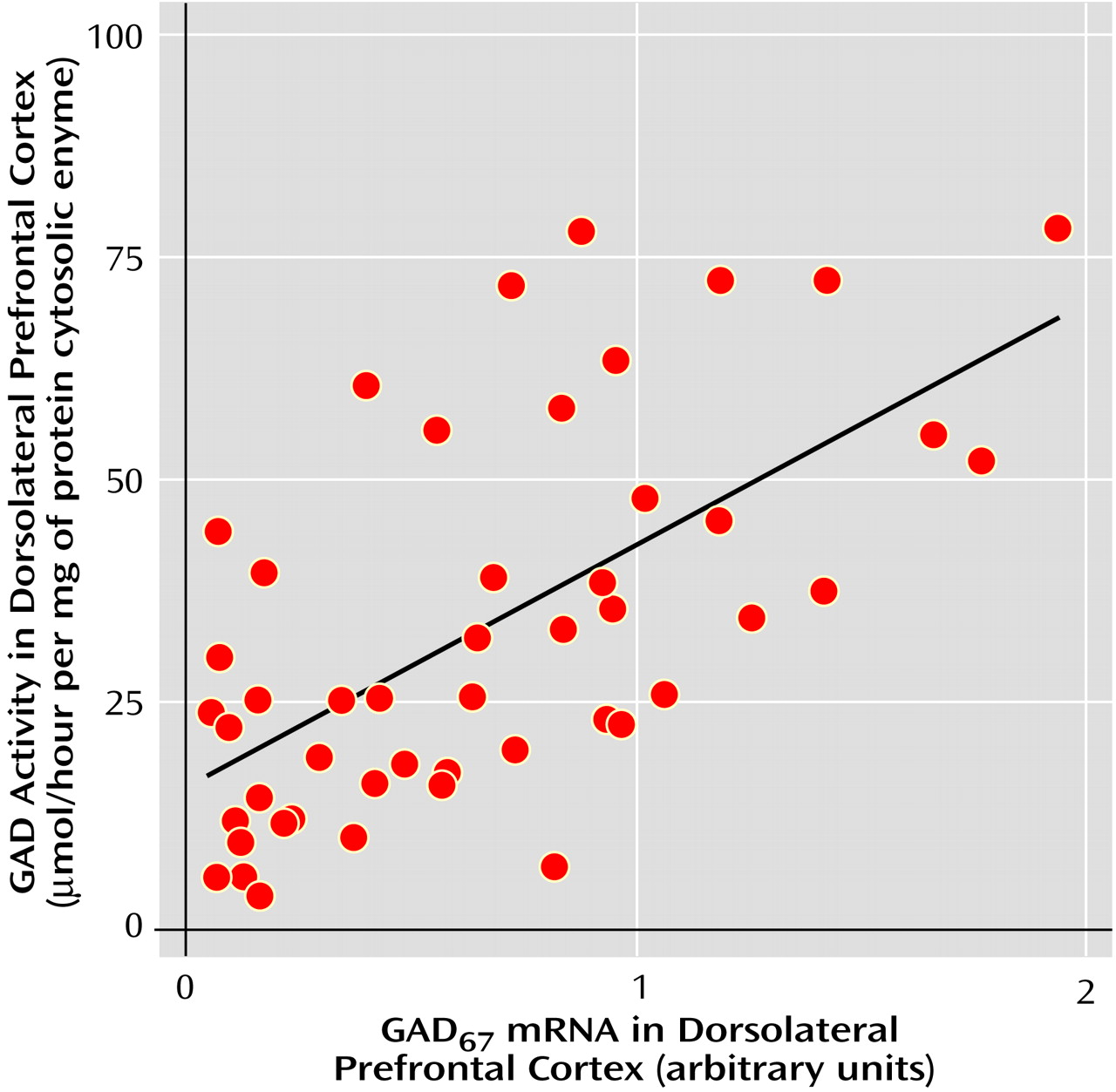
Rates for glutamic acid decarboxylase, GABA-transaminase, and succinic semialdehyde dehydrogenase activity are shown in
Table 2. There were no significant activity differences in GABA-transaminase and succinic semialdehyde dehydrogenase between schizophrenic and normal comparison subjects, whether the enzyme activities were expressed as units/mg of protein or per unit of citrate synthase activity. In contrast, there was significantly greater glutamic acid decarboxylase activity in the schizophrenic subjects (F=3.61, df=2, 46, p<0.04) (Newman-Keuls test of normal comparison versus schizophrenic subjects, p=0.04) (154%) and in normal comparison versus age-matched schizophrenic subjects (t=–3.4, df=18, p=0.03) (206%). Differences in glutamic acid decarboxylase activities in the dorsolateral prefrontal cortex of identical specimens from Alzheimer’s disease patients were not observed (94%). Because differences in the agonal state have been linked to variations in brain glutamic acid decarboxylase activity, it is important to note that tissue pH was not significantly different among the three groups (F=0.39, df=2, 46, p=0.67).
The activities of α-ketoglutarate dehydrogenase, glutamate dehydrogenase, glutamine synthetase, and AST and ALT in the dorsolateral prefrontal cortex did not differ significantly (p>0.10) in comparisons of schizophrenic and normal comparison subjects as whole groups or when matched for age, whether the enzyme activities were expressed per mg of protein or per unit of citrate synthase activity (
Table 2).
There was statistically significantly greater phosphate-activated glutaminase activity in the dorsolateral prefrontal cortex (
Table 2) in schizophrenic versus normal comparison subjects when the entire groups were assessed (450%) (F=4.85, df=2, 46, p<0.02; Newman-Keuls test of comparison versus schizophrenic subjects, p=0.04) or when they were assessed on an age-matched basis (438%) (t=–2.3, df=18, p<0.04).
Phosphate-activated glutaminase activity was virtually abolished (95%–100%, data not shown) in the presence of 2.0 mM of 6-diazo-5-oxo-
l-norleucine, a selective inhibitor of phosphate-activated glutaminase
(46), indicating that the observed activities derived from phosphate-activated glutaminase. To test for the disease specificity of elevated phosphate-activated glutaminase activities in the dorsolateral prefrontal cortex of schizophrenic subjects, the level of phosphate-activated glutaminase activity was determined in the dorsolateral prefrontal cortex of the Alzheimer’s disease group. Phosphate-activated glutaminase activity in the group with Alzheimer’s disease was not significantly different from that in the normal comparison subjects (Newman-Keuls test, p=0.80), but it differed significantly from the schizophrenia group (Newman-Keuls test, p=0.03). To determine whether the differences in phosphate-activated glutaminase activity in the schizophrenia group were specific to the dorsolateral prefrontal cortex, the activities of phosphate-activated glutaminase in the primary visual cortex (Brodmann’s area 17) were compared in the same groups of schizophrenic and normal comparison subjects. In the visual cortex, there was significantly less phosphate-activated glutaminase activity than in the dorsolateral prefrontal cortex in both schizophrenic and comparison subjects (24.9% of the dorsolateral prefrontal cortex) (t=13.9, df=37, p=0.0007). Although whole-group phosphate-activated glutaminase activity in the visual cortex of the schizophrenic subjects was nominally greater than that in the visual cortex of the comparison subjects, this difference was not statistically significant (Newman-Keuls test, p=0.10). The activity of phosphate-activated glutaminase did not correlate significantly with postmortem interval, age, tissue pH, weeks free from neuroleptic exposure before death, or storage time (r=–0.16 to 0.24, N=40, p>0.10).
There were significant correlations between glutamic acid decarboxylase and phosphate-activated glutaminase activities in the dorsolateral prefrontal cortex of the entire study group (r=0.78, N=40, p=0.00001) as well as in the schizophrenic group when it was assessed alone (r=0.80, N=27, p=0.00001). In contrast, the activities of glutamic acid decarboxylase and phosphate-activated glutaminase did not correlate significantly with each other when assessed in the normal comparison group alone (r=0.26, N=13, p=0.38), which suggests that the significant correlation between phosphate-activated glutaminase and glutamic acid decarboxylase activity in the group as a whole was predominantly attributable to the relationship between glutamic acid decarboxylase and phosphate-activated glutaminase in the schizophrenia group.
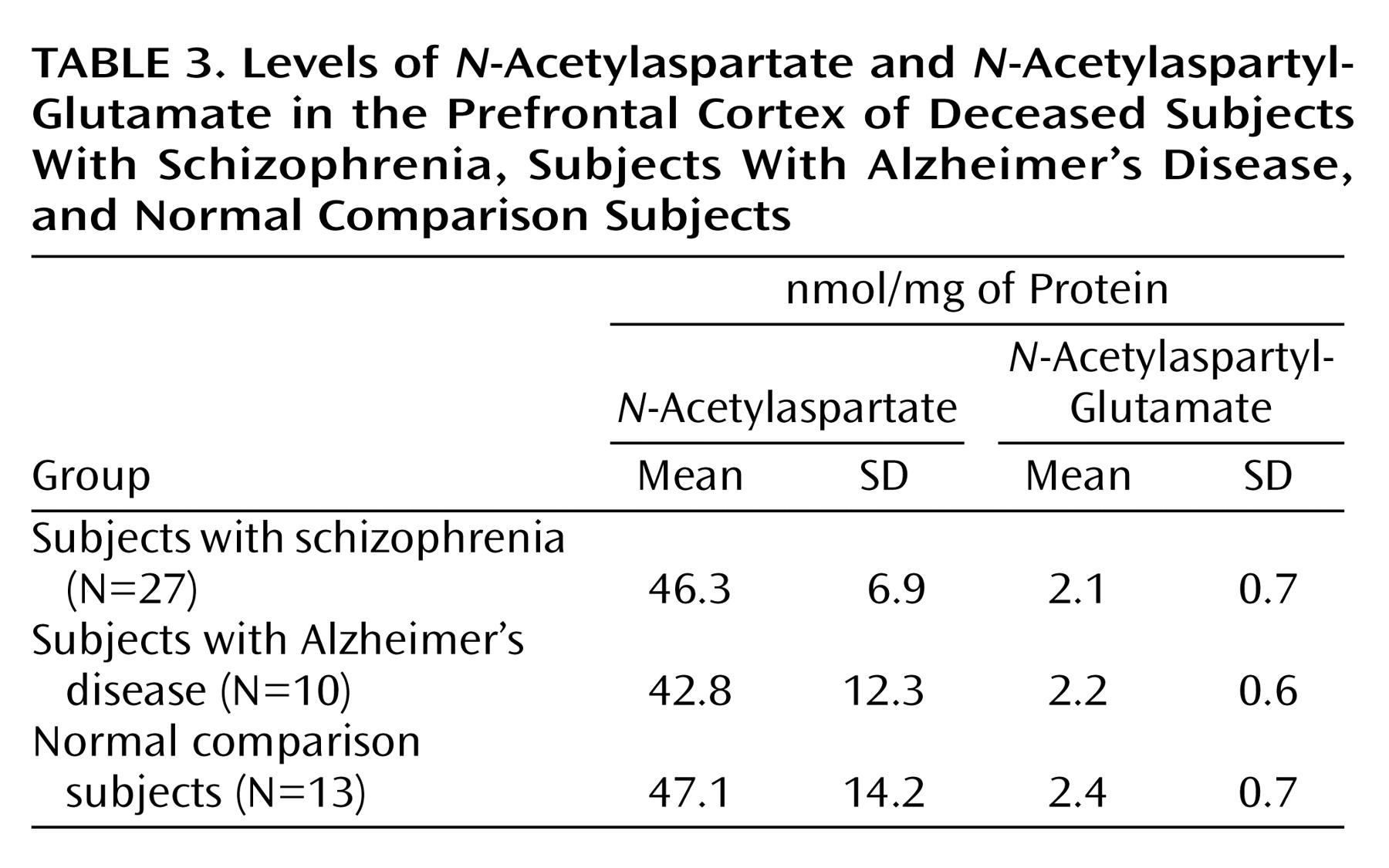
N-Acetylaspartate and
N-acetylaspartyl-glutamate levels in the dorsolateral prefrontal cortex of the subjects with schizophrenic, the comparison subjects with Alzheimer’s disease, and the nonpsychiatric comparison subjects are shown in
Table 3. No statistically significant differences in
N-acetylaspartate or
N-acetylaspartyl-glutamate levels were observed among the groups. The ratios of
N-acetylaspartate to
N-acetylaspartyl-glutamate levels were approximately 14:1, consistent with previous findings
(3).
The activities of phosphate-activated glutaminase and glutamic acid decarboxylase did not correlate significantly with the number of weeks that each schizophrenic subject had been free of exposure to neuroleptic drugs (r=0.10 to –0.01, N=40, p>0.60). Additionally, when the schizophrenic group was stratified into patients who had been exposed to neuroleptics until the time of death (N=15) versus those who had been neuroleptic free for 6 weeks or more (N=9), the activities of phosphate-activated glutaminase and glutamic acid decarboxylase did not differ significantly between the groups (t<0.8, df=22, p>0.43).
Differences in cortical phosphate-activated glutaminase activities in rats subchronically treated with haloperidol relative to normal control subjects receiving saline vehicle were not evident (haloperidol: N=6, mean=0.034 activity units/units of citrate synthase activity, SD=0.005; saline vehicle: N=6, mean=0.036 activity units/units of citrate synthase activity, SD=0.005) (t=–0.8, df=10, p=0.44). Similarly, cortical glutamic acid decarboxylase activity was not altered by haloperidol treatment (haloperidol treatment: N=6, mean=0.268 activity units, SD=0.080; saline-vehicle treatment: N=6, mean=0.256 activity units, SD=0.110) (t=1.1, df=10, p=0.30).