Rigorous scientific examination has identified selective serotonin reuptake inhibitors (SSRIs) as the first-line treatment for clinically significant premenstrual syndromes (PMS)
(1–
16), particularly its severe form of premenstrual dysphoric disorder (PMDD) as described in DSM-IV. Observations of the rapid response to SSRIs when administered in a continuous dosing regimen for severe PMS or PMDD resulted in further investigation of their use in the symptomatic premenstrual weeks of the menstrual cycle. Within the last several years, a number of small studies and two large multicenter trials reported efficacy for premenstrual dosing in patients with PMDD
(17–
22). Although the pharmacologic reasons for the swift response of PMS patients to SSRIs have not been demonstrated, evidence of the potential role of altered serotonergic transmission is increasing
(23). Brain serotonin transmission is influenced by alterations of plasma concentrations of estradiol and progesterone
(24,
25), suggesting a possible pathway by which the gonadal steroids may be involved in this menstrual cycle-related disorder
(26).
While studies have shown that both continuous and intermittent dosing with an SSRI are effective for severe PMS/PMDD, there has been little direct comparison to determine whether one of the dosing regimens should be preferred in clinical practice. One study concluded that premenstrual citalopram dosing was more effective than the continuous dosing regimen
(11), but differences were marginal, and the small groups had low statistical power. Whether premenstrual dosing is more effective than continuous dosing (or vice versa) in reducing premenstrual symptoms or the side effects of medication has remained an open question.
The objectives of this study were to compare the efficacy and acceptability of continuous versus intermittent SSRI treatment in women with severe premenstrual syndrome with the hypothesis that both SSRI regimens would be more effective than placebo. On the basis of preliminary data
(5,
27), we further hypothesized that women with higher symptom levels in the follicular phase of the cycle (a possible indication of subclinical levels of depression) would respond to full-cycle dosing but not luteal-phase dosing and that a history of depression would not be associated with treatment response.
Method
Patient Selection
The study was conducted at the Premenstrual Syndrome Program of the University of Pennsylvania Medical Center from 1998 to 2002. The study was approved by the Institutional Review Board of the University of Pennsylvania, and subjects provided written informed consent.
Inclusion criteria were age 18–45 years; regular menstrual cycles of 22–35 days; a positive urine test indicating probable ovulation; persistent premenstrual symptoms for at least 6 months; global report of moderate to severe impairment in work, family life, or social activity; general good health; no major psychiatric diagnosis within the past year, and meeting the stated PMS criteria with confirmation by prospective daily symptom ratings. The Daily Symptom Rating Form
(28) was used and included the following symptoms: depression, feeling hopeless or guilty, anxiety/tension, mood swings, irritability/anger, decreased interest, concentration difficulties, fatigue, food cravings/increased appetite, insomnia or hypersomnia, feeling out of control/overwhelmed, poor coordination, headache, aches, swelling/bloating/weight gain, cramps, and breast tenderness.
Exclusion criteria were any major axis I psychiatric diagnosis, including major depression, currently or within the past year as assessed by the Structured Clinical Interview for DSM-IV Axis I Disorders (SCID)
(29); use of psychotropic medications that could not be discontinued for the duration of the study; use of any prescription, nonprescription, herbal, or other therapies for PMS; pregnancy; currently breast-feeding; hysterectomy; symptomatic endometriosis; irregular menstrual cycles; not using medically approved contraception; serious health problems; risk of suicide; or alcohol or drug abuse within the past year.
Symptom Criteria
The diagnosis of severe PMS was based on prospective rating of the previously validated Daily Symptom Rating Form
(28) and patient global ratings of functioning. Subjects rated functioning in work, family life, social activity, and sexual activity on 5-point scales ranging from 0 (no disruption) to 4 (severe disruption). A rating ≥2 on one or more of the function scales was required.
Subjects rated symptoms daily with the Daily Symptom Rating Form on a 5-point scale (0=none, 4=extreme). Scores were calculated by adding the ratings of cycle days 5 through 10 for the postmenstrual scores (day 1 was the first day of menses) and by adding the ratings for the 6 days before menses for the premenstrual scores. The Daily Symptom Rating Form criteria for severe PMS required a total premenstrual Daily Symptom Rating Form score ≥80 and an increase of ≥50% over the postmenstrual score for the mean of the three screen cycles and for the single-blind placebo cycle.
In addition to meeting the criteria for severe PMS, 60% of the subjects met DSM-IV criteria for PMDD for which a stringent definition was used: Daily Symptom Rating Form ratings of 3 or 4 on at least two premenstrual days for at least five PMDD symptoms, with the same symptoms rated 0 or 1 (absent or minimal) postmenstrually. On average, the subjects who did not meet PMDD criteria had three symptoms that strictly met the PMDD criteria.
We studied severe PMS because of its clinical importance inasmuch as many women seek medical treatment but do not meet the stringent PMDD criteria, particularly the requirement of five specific PMDD symptoms. The strong overlap between severe PMS and PMDD is evident in our data. Moreover, several recent population-based studies of the prevalence of premenstrual symptoms underscored the importance of “near-threshold” cases
(30,
31). Such cases failed to have the requisite number of PMDD symptoms but were nonetheless highly correlated with functional impairment, a widely recognized indicator of the need for treatment.
Women requesting treatment for premenstrual symptoms were screened briefly by telephone and were provided with the Daily Symptom Rating Form with instructions for prospective daily rating. Potential subjects made the first full screening visit following menses (cycle days 6–12) (N=555) to assess the postmenstrual symptom levels. At this visit, the Daily Symptom Rating Form was reviewed, and the SCID was administered. Subjects were given a urine test kit with instructions for performing the at-home test to indicate probable ovulation. The second screening visit was scheduled in the premenstrual week of the same cycle to assess premenstrual symptom status. Physical and pelvic examinations were conducted and specimens for laboratory screening (complete blood cell count and blood chemistry panel) were obtained. Women who remained eligible were given two bottles of placebo tablets (single-blind) and instructed to take one tablet daily throughout the third screening cycle. Subjects who continued to meet the eligibility criteria were randomly assigned to a double-blind treatment condition for three menstrual cycles.
Design
This was a stratified, randomized, double-blind, parallel treatment trial that compared full-cycle and premenstrual dosing regimens of sertraline with a placebo. Stratification was used to assure equal distribution of the variables that were hypothesized to affect treatment response. The hypotheses were based on preliminary findings that postmenstrual symptom severity before treatment was highly predictive of subsequent Daily Symptom Rating Form scores
(27) and that a history of depression may potentially affect response to an SSRI, although the latter has not been identified in PMS treatment studies. Random allocation to the study groups was generated from random-number tables before the start of the study by a technician with no clinical contact. Group size requirements were determined from analysis of Daily Symptom Rating Form data in a previous trial (5), using assumed differences (N=30, N=40, N=45), standard deviation (SD=60, SD=70) and correlations between baseline and endpoint scores (0.20, 0.30, 0.40) and indicated that a sample of 135 evaluable subjects (45 in each treatment arm) would provide power greater than 0.90 with alpha at 0.05 and the inclusion of up to four covariates.
Study Doses
Sertraline and placebo tablets were identical in appearance. All subjects received two bottles of medication at each visit with the same code number assigned at randomization used throughout the double-blind treatment. All subjects took tablets daily, starting with Bottle A on day 3 of menses, switching to Bottle B at 14 days before the expected menses and continuing through day 2 of menses. Starting at day 3, subjects received either 50 mg sertraline (the daily dosing group) or placebo. At 14 days before menses, subjects in the premenstrual dosing group were switched from placebo to 50 mg sertraline. The initial dose for all subjects was 1 tablet per day continued through day 2 of the next cycle. In the absence of clear improvement or dose-limiting side effects, the dose was raised to two tablets per day (100 mg sertraline or placebo) in the second or third double-blind treatment cycles. Dose increases started with bottle 2 at day 14. Subjects who increased to two tablets during the second cycle continued with two tablets through the third cycle unless precluded by side effects. To maintain the study blind, all subjects taking two tablets per day at endpoint were tapered to one tablet per day for 7 days before stopping medication. At each visit, subjects returned all unused medication, which was counted by the study coordinator, and received medication for the next cycle. The mean dose levels in the third treatment cycle were 74 mg/day (SD=25) in the full-cycle sertraline group, 76 mg/day (SD=25) in the luteal-phase sertraline group, and a dose equivalent for the placebo tablets of 85 mg/day (SD=23) (F=2.30, df=2, 114, p=0.11).
Outcome Measures
The primary outcome measure was the total score on the premenstrual Daily Symptom Rating Form, which was calculated from the daily symptom ratings that were maintained throughout the study. The five statistically derived Daily Symptom Rating Form factors and the individual Daily Symptom Rating Form items were also examined. The Daily Symptom Rating Form factors included mood (irritability/anger, mood swings, anxiety/tension, depression, feeling out of control, feeling worthless/guilty, or decreased interest); behavior (poor coordination, insomnia, difficulty concentrating/confusion, or fatigue); pain (aches, headache, or cramps); physical symptoms (breast tenderness or swelling/bloating); and food cravings/increased appetite (a single item in the factor analysis). The second outcome measure was the Subject Global Ratings of Functioning (family life, work, social activity, sexual function). Each dimension was rated on a 5-point scale ranging from 0 for no disruption to 4 for severe disruption. Improvement was defined as a positive change (range=1–4 points) from baseline at treatment endpoint.
Statistical Analysis
Analysis of the Daily Symptom Rating Form scores was conducted for all observations and for the last observation carried forward by using an intent-to-treat approach that was defined before the study as including all subjects with at least one treatment response rating. A mixed analysis of variance (ANOVA) model for repeated measures with unstructured covariance was used. The model for the stratified study design included treatment (three groups), baseline symptoms (average premenstrual Daily Symptom Rating Form scores for the three screen cycles), treatment cycle (three), postmenstrual symptom severity before treatment (high, low), and history of major depression (yes, no) and examined the premenstrual Daily Symptom Rating Form scores over the 3 treatment months. Interactions between treatment and the covariates were examined. Results are reported for all observations and for the last observation carried forward as identified. Results of the mixed model were also examined by using the general linear model procedure to test the change from baseline of the mean Daily Symptom Rating Form premenstrual scores for three treatment cycles and the results at endpoint. The general linear model analyses were consistent with the reported results. Before the multivariate analyses, the data were checked for distributions and influential outliers. To further determine possible effects of outliers and the consistency of results, logged values of the treatment data were examined and agreed with the results reported here. A completer analysis was also consistent with the reported results. Comparisons of sample characteristics at baseline were examined by using ANOVA for continuous variables or frequency distributions with the chi-square test for categorical variables. The analysis of the global functioning ratings examined the frequency distributions of the change from baseline at treatment endpoint. Statistical results with p≤0.05 for a two-tailed interpretation were considered statistically significant. The statistical software package was SAS, Version 8.0 (SAS Institute Inc., Cary, N.C.).
Results
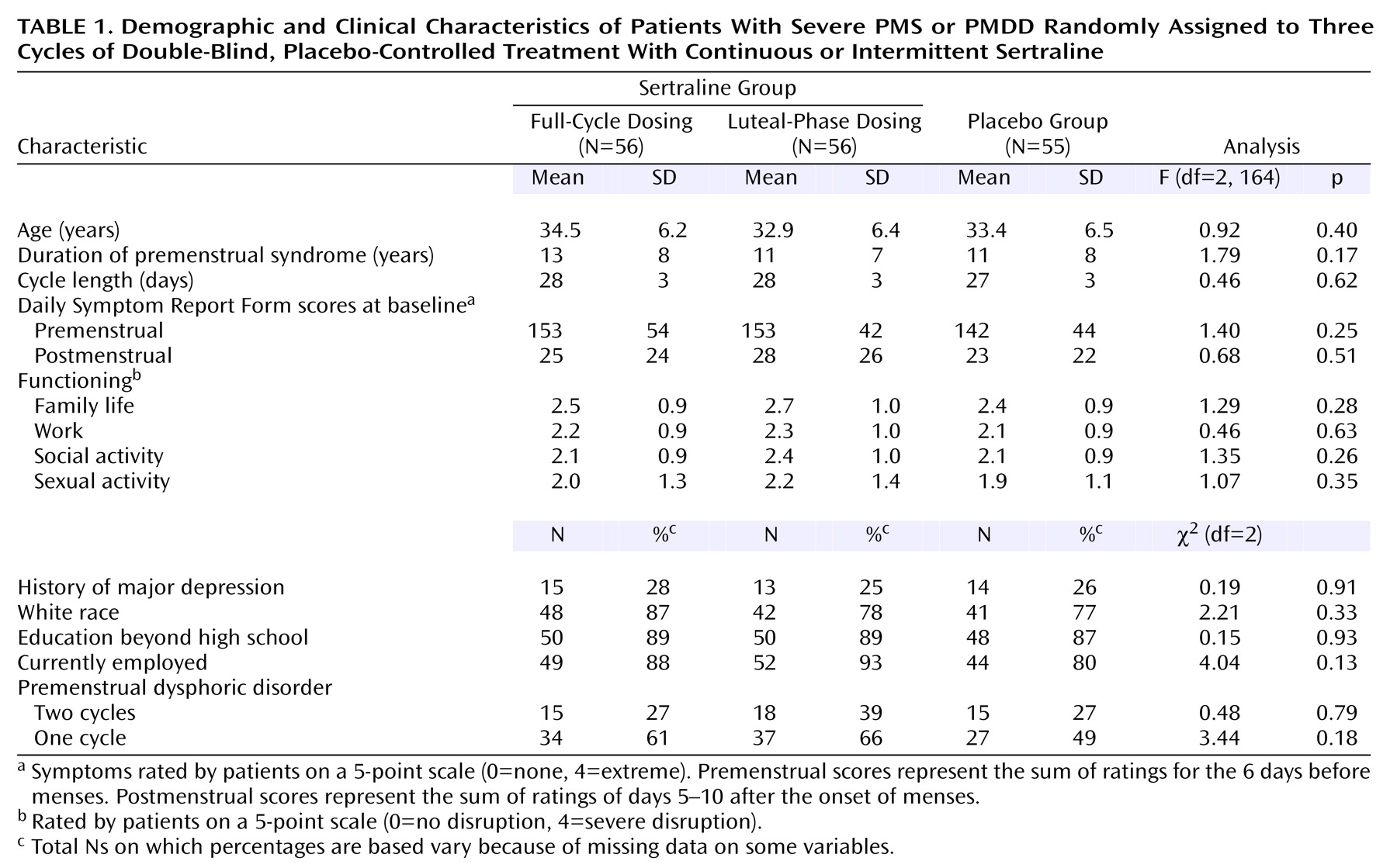
Subject characteristics were similar in the three treatment groups (
Table 1).
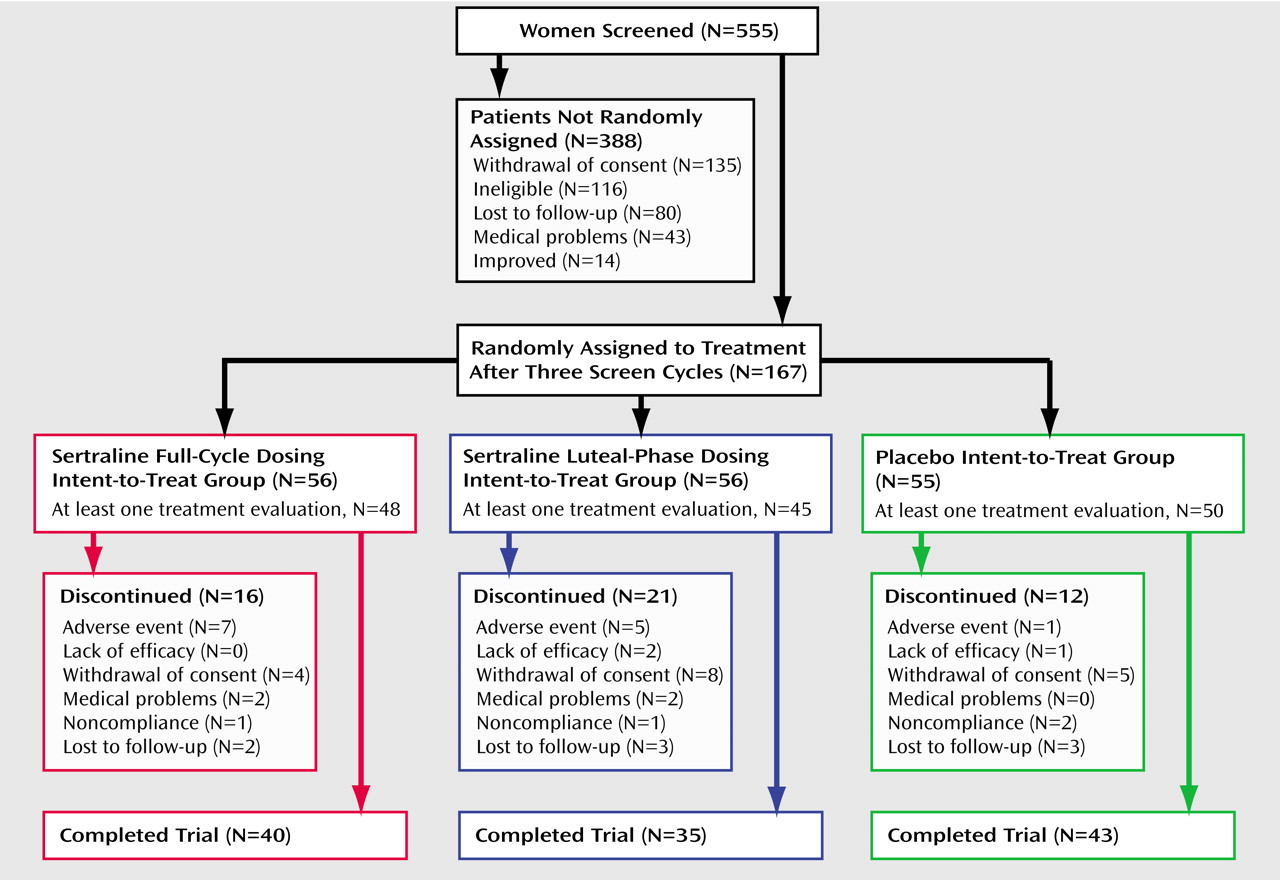
Figure 1 summarizes the progress of the subjects through the study. One hundred sixty-seven subjects were randomly assigned to a treatment condition. Twenty-four subjects discontinued before providing any treatment response data, and another 25 subjects discontinued after providing at least one treatment response. Differences between treatment groups were not significant (χ
2=3.32, df=2, p=0.19). The numbers and reasons for discontinuation are shown in
Figure 1. Comparisons of the baseline data in
Table 1 between the subjects with no treatment response data and the remaining subjects showed that the group with no treatment data were more likely to be minority subjects (29% minority versus 11% white) (χ
2=6.24, df=1, p=0.01) and had higher baseline postmenstrual Daily Symptom Rating Form scores (t=1.96, df=165, p=0.05).
Adverse Events
Adverse events were reported at each visit in response to general clinical questioning by a clinician and a subject self-report questionnaire. Subjects also were provided a medication problem checklist to record at home any side effects experienced while taking medication from Bottle A in order to identify possible discontinuation symptoms when the premenstrual dosing group shifted from sertraline to placebo in each treatment cycle. There were no serious adverse events that required medical intervention and no reports of withdrawal symptoms that required medication adjustments. Eight percent of the subjects (N=13 of 167) randomly assigned to a treatment condition indicated that adverse events were the reason for withdrawing from the study (full-cycle sertraline group: N=7; luteal-phase sertraline group: N=5; placebo group: N=1) (χ2=4.45, df=2, p=0.10).
The most frequent adverse events were gastrointestinal (19%), decreased libido or orgasm (15%), headache (14%), insomnia (13%), dry mouth (13%), nausea (13%), and nightmares (12%). Adverse events diminished with time and adjustment to the medication, particularly in the full-cycle dosing group, where reports did not differ from the placebo group in the third treatment month (χ2=0.77, df=1, p=0.38). The luteal-phase dosing group, which stopped and restarted the active medication each month, continued to report more adverse events compared with placebo in the third treatment month (χ2=4.8, df=1, p=0.03).
Treatment Response
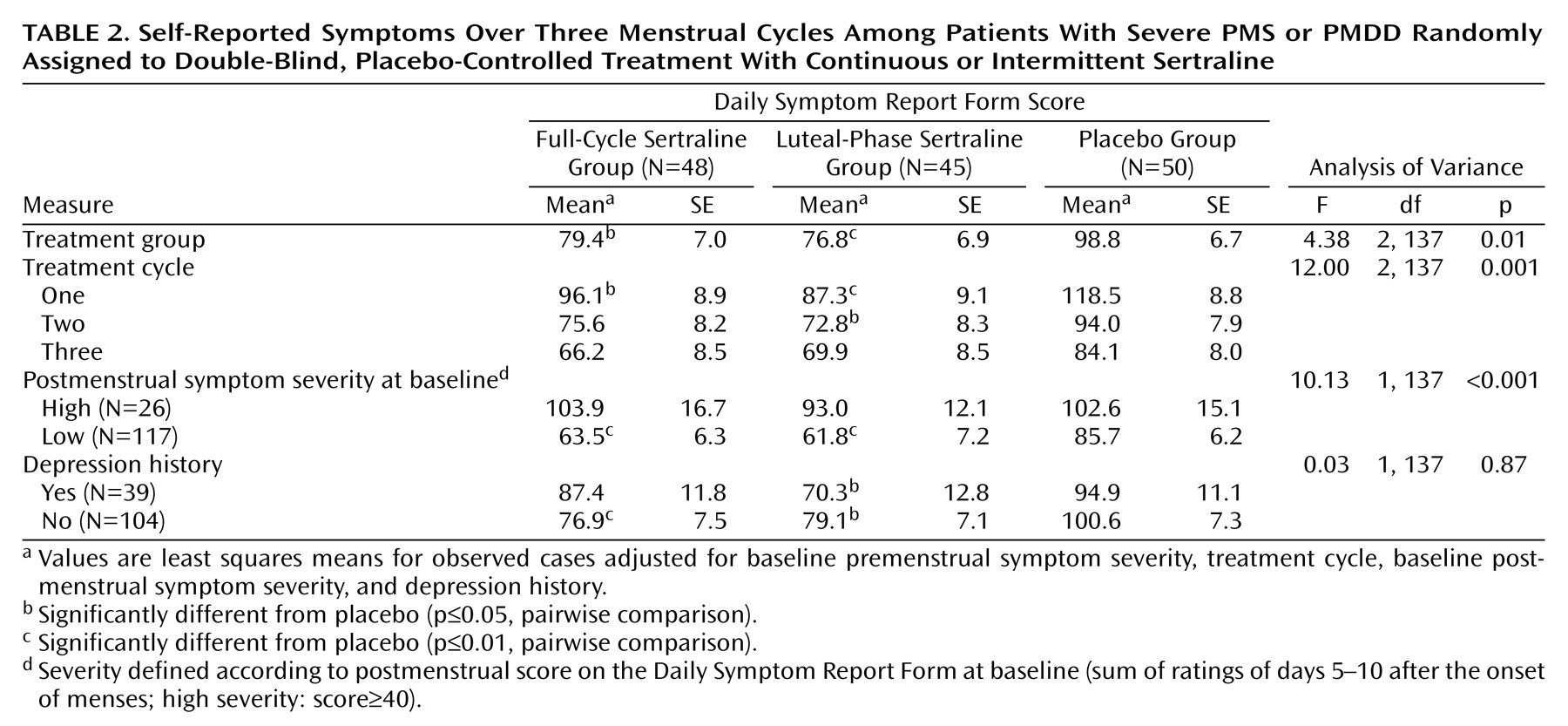
Improvement from baseline for all observed cases (intent to treat) as assessed by the premenstrual Daily Symptom Rating Form scores over three treatment cycles was significant (F=4.38, df=2, 137, p<0.01) (
Figure 2,
Table 2). Both sertraline groups improved significantly more than the placebo group (full-cycle dosing versus placebo: t=–2.39, df=137, p=0.02); luteal-phase dosing versus placebo: t=–2.65, df=137, p=0.009). There was little difference between the two sertraline groups (t=0.31, df=137, p=0.76). There were no significant interactions of treatment with cycle (F=0.70, df=4, 137, p=0.59), postmenstrual symptom group (F=0.38, df=2, 135, p=0.68), or history of depression (F=0.29, df=2, 135, p=0.75).
Improvement from baseline as assessed by the premenstrual Daily Symptom Rating Form scores over three treatment cycles with last observation carried forward was significant (F=3.83, df=2, 137, p=0.02). The sertraline-treated groups showed greater improvement than the placebo group (full-cycle dosing versus placebo: t=–1.93, df=137, p=0.055; luteal-phase dosing versus placebo: t=–2.67, df=137, p=0.009). There was no significant difference between the two sertraline groups (t=0.78, df=137, p=0.44).
An analysis of completers of the three treatment cycles also showed significant improvement from baseline over the three treatment cycles as assessed by the premenstrual Daily Symptom Rating Form scores (F=4.61, df=2, 111, p≤0.01, data not shown). In the completer group, both full-cycle (t=–2.80, df=111, p=0.006) and luteal-phase (t=–2.28, df=77, p<0.03) sertraline dosing were significantly better than placebo.
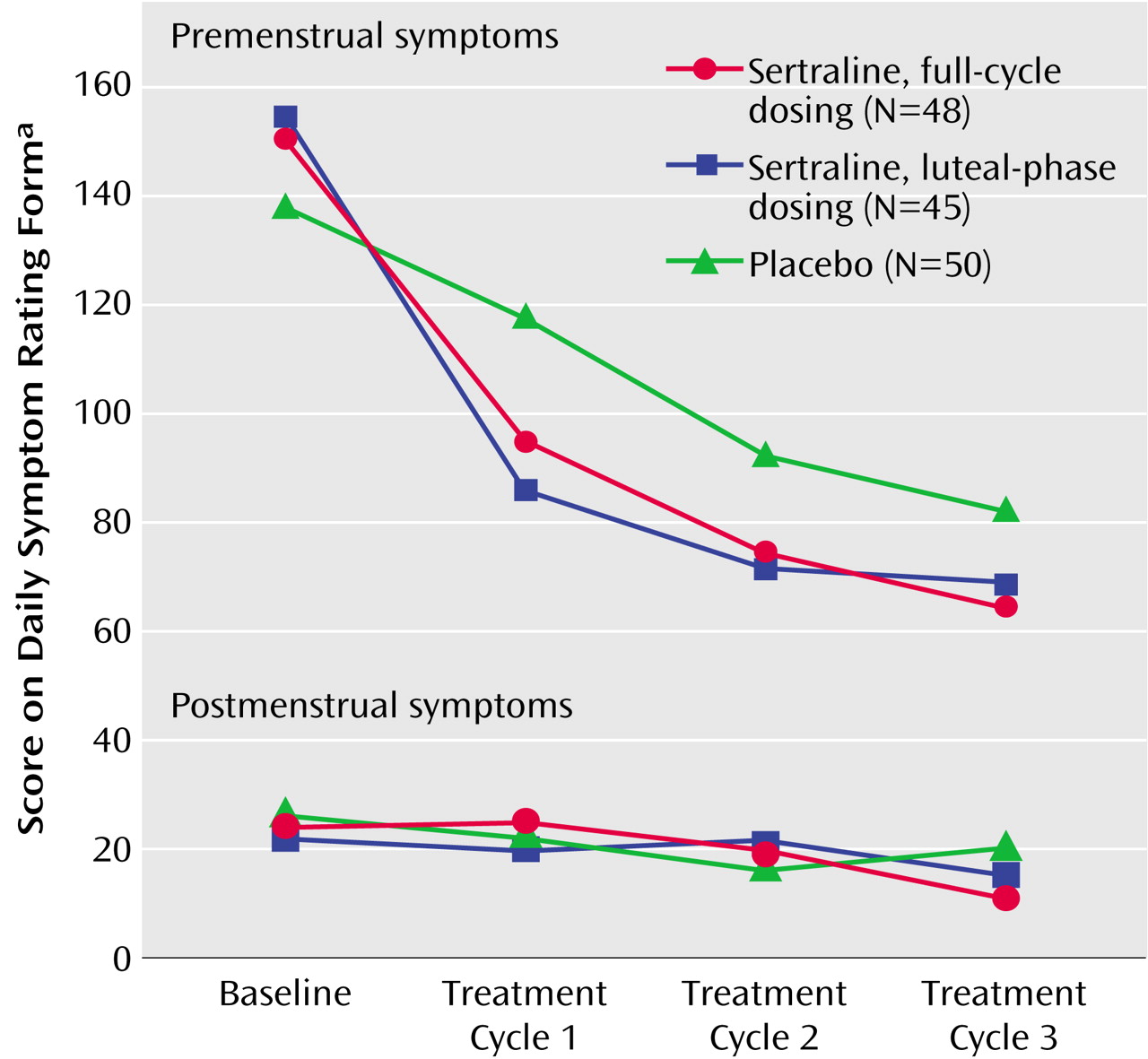
Inspection of the mean Daily Symptom Rating Form scores by cycles showed that the greatest decrease in Daily Symptom Rating Form scores from baseline was in the first month of double-blind treatment: 42% in the full-cycle group and 45% in the luteal-phase dosing group compared with 26% in the placebo group (unadjusted mean Daily Symptom Rating Form scores). However, improvement in the placebo group continued gradually and did not significantly differ from the two sertraline groups in the last treatment cycle as shown in
Figure 2 (sertraline versus placebo at endpoint: t=–1.59, df=138, p=0.11).
Effect of postmenstrual symptom severity
The random allocation to treatment was stratified for high or low postmenstrual symptom levels using a previously identified mean postmenstrual Daily Symptom Rating Form score of 40 for the cutoff point
(27). Treatment response as assessed by total premenstrual Daily Symptom Rating Form scores was significantly greater in the patients with low postmenstrual symptoms (F=10.13, df=1, 137, p=0.002). Although both the high and low postmenstrual symptom groups improved, the low postmenstrual symptom group reached a clinically acceptable level of improvement, whereas the high postmenstrual symptom group, which started at a higher symptom level, remained more symptomatic with Daily Symptom Rating Form scores remaining well above the entry criterion (
Table 2).
Effect of depression history and diagnosis
The stratification variable of history of major depression had no association with treatment response (
Table 2). Treatment results remained the same when diagnosis (severe PMS or PMDD) was added to the model (treatment F=4.30, df=2, 136, p<0.02). Diagnosis of severe PMS versus PMDD was not significant (F=1.22, df=1, 136, p=0.27).
Symptom, Function, and Clinical Response
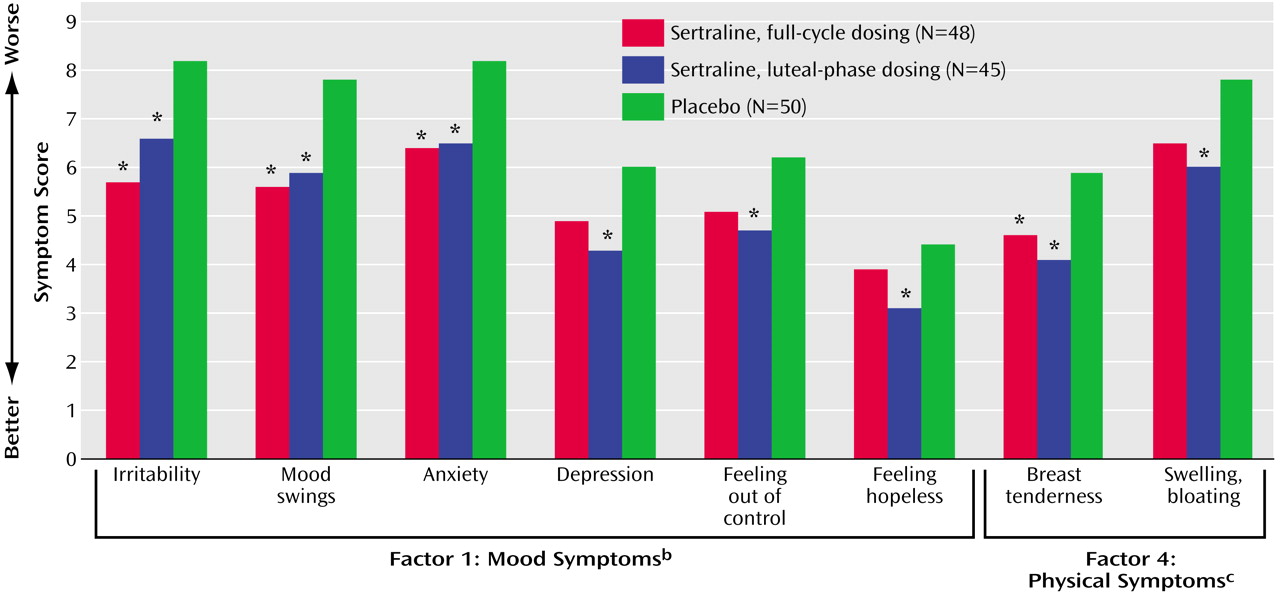
Analysis of the Daily Symptom Rating Form factor scores showed that the sertraline-treated groups consistently showed more improvement on all factors compared with the placebo group, with a statistically significant treatment effect for factor 1, mood symptoms, and factor 4, physical symptoms (
Figure 3).
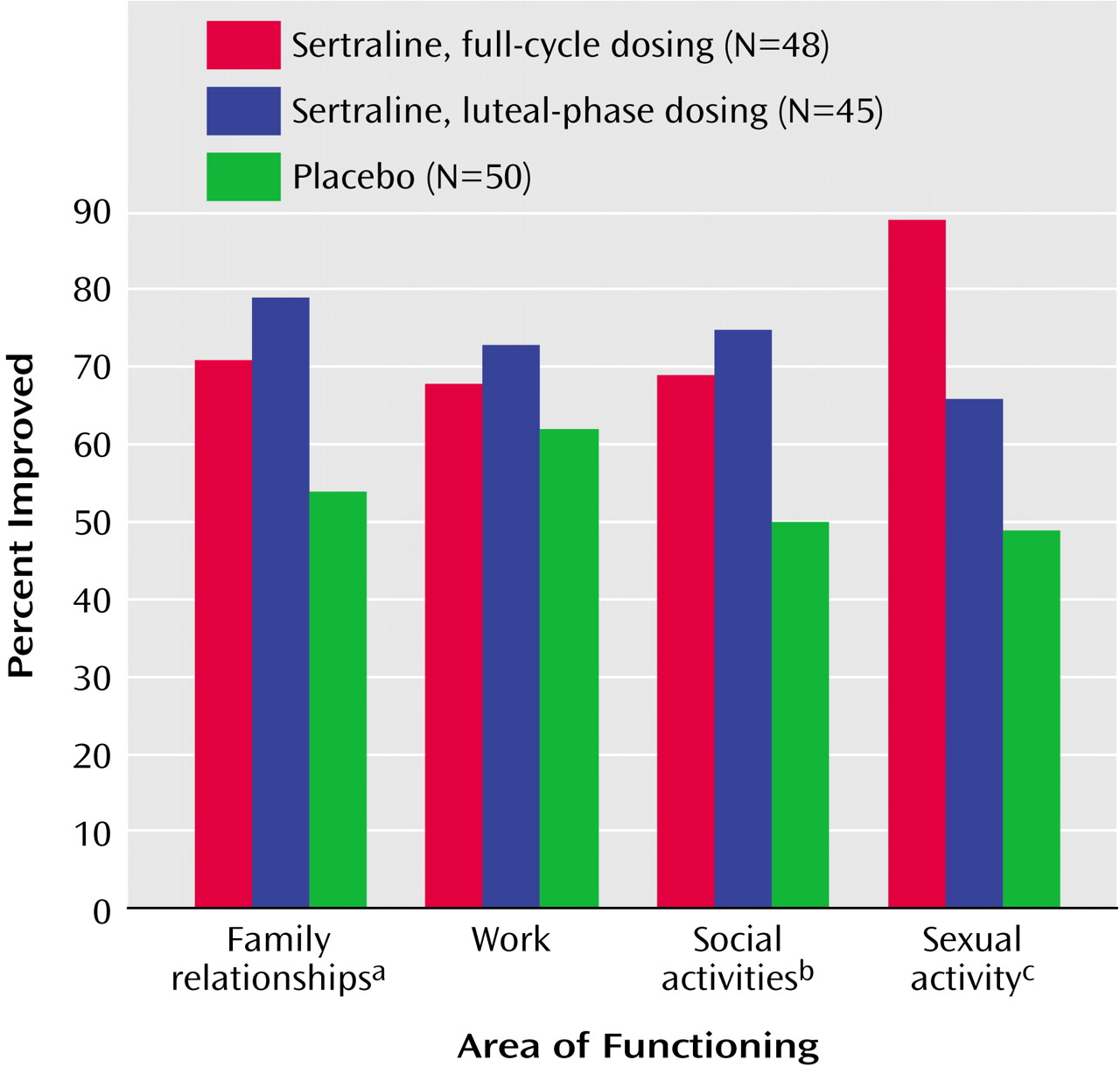
Functional impairment was a criterion for study enrollment and was similar in the three treatment groups at baseline (shown in
Table 1). Significantly more subjects in the sertraline-treated groups than the placebo group reported improvement in family relationships, social activities, and sexual functioning as assessed by the change from baseline in subjects’ global ratings at endpoint (
Figure 4).
The clinical response rate (defined as the subjects with ≥50% reduction in the mean premenstrual Daily Symptom Rating Form scores over the three treatment cycles from the pretreatment baseline) was 63% (N=30 of 48) in the full-cycle group, 51% (N=23 of 45) in the luteal-phase dosing group, and 36% (N=18 of 50) in the placebo group (χ2=6.94, df=2, p=0.03). A 75% mean treatment reduction from baseline in total premenstrual Daily Symptom Rating Form scores was reported by 19% of the full-cycle group, 22% of the luteal-phase dosing group, and 6% of the placebo group (p=0.06, Fisher’s exact test).
Discussion
This comparison of full-cycle versus premenstrual sertraline treatment for severe PMS showed no significant differences between the two dosing regimens. While both sertraline-treated groups improved significantly more than the placebo group, response did not differ between the two sertraline dosing groups in any treatment cycle. Adverse events also did not differ between the two sertraline groups, although fewer adverse events were reported in the full-cycle dosing group after 3 months of treatment. However, premenstrual dosing clearly offers less exposure to drug, which is an important factor to consider in the risk-benefit equation. The conclusion from these data is that the decision between full-cycle and premenstrual dosing for women with severe PMS or PMDD and no other comorbid diagnoses can be based on patient/physician preference and individual experience of side effects.
Treatment response occurred primarily in the first month of treatment, as has been consistently observed in other clinical trials of SSRIs for PMS treatment
(4–
8). The placebo-treated group evidenced a slower course of improvement, which remained less improved but not significantly different from the sertraline-treated groups at endpoint. This placebo response was higher than in our previous PMS trials
(5,
8,
32) but notably similar to other placebo response rates in several large PMDD trials in this same time period
(4,
21). We speculate that, in addition to the supportive care of the treatment trial, the recent Food and Drug Administration approvals of fluoxetine and sertraline for PMDD treatment and the greatly increased use of these medications in medical treatments raised
expectations of their efficacy for PMS, which was then reflected in the subjects’ experience of treatment, whether or not it was active drug. The results clearly demonstrate the more rapid efficacy of medication but also indicate the importance of nondrug factors in clinical care
(33,
34) and suggest the potential for cognitive therapy
(35–
38) or supportive treatments as options for severe PMS.
The salient indicator of severe PMS is that symptoms are minimal or absent following menses, but there is no standard measure for operationalizing the maximum postmenstrual symptom levels or the minimum premenstrual severity required for a PMS or a PMDD diagnosis. The present results showed that higher postmenstrual symptom levels at baseline predicted a poorer response to treatment. This confirmed our previous findings
(27), although the small number of subjects with high postmenstrual symptom levels in this protocol limits conclusions.
The results failed to support the hypothesis that subjects with higher postmenstrual symptom levels would respond better to full-cycle dosing, inasmuch as
both drug-treated groups with high postmenstrual scores responded poorly. Possibly the higher postmenstrual symptom levels were an indication of undiagnosed depression, but all subjects in the high postmenstrual group had postmenstrual scores on the 17-item Hamilton Depression Rating Scale in the normal range (<7), providing little evidence of current depression. Five of these women had a history of major depressive disorder, and five had a history of minor depression, but they responded no better to the daily dosing regimen in this protocol. We speculate that the poorer response is due in part to persistent personality characteristics, which are not menstrual-cycle specific but are associated with symptom ratings, although such associations are not clearly defined at this time
(39,
40). Most important, the present results provide further evidence of the need to clearly define how the symptom criteria for severe PMS and PMDD are operationalized, as recently demonstrated by Smith et al.
(41).
A history of depression as determined in SCID interview was identified in 27% of the subjects but was not associated with treatment response. This adds support to other observations that suggest that PMS is a distinct disorder with underlying mechanisms that differ from other depressive disorders
(3,
5,
7).
Although the duration of this study is usual for PMS trials, the 3-month treatment interval does not answer several important clinical questions. Whether response to medication is maintained in long-term treatment and whether there are differences between continuous and intermittent dosing regimens in long-term treatment are not known. Possibly subjects with limited responses would improve with further dose increases, although data thus far show very little incremental improvement at higher dose levels for PMS or PMDD
(2,
5). Although adverse events did not differ between the two sertraline groups but appeared more likely to subside in the continuous dosing group, a much larger sample is needed to definitively determine any differences in adverse events between the dosing regimens. The sample did not include patients with comorbid physical or psychiatric disorders, which are frequently exacerbated premenstrually
(42). Comorbidity remains an important and unstudied aspect of treatment for PMS/PMDD. Finally, it is important to consider that although approximately 60% of the sertraline-treated women responded well, about 40% did not. While this is a very high response rate and consistent with all other reports of SSRI treatments for PMS/PMDD, further evidence-based information about treatments and underlying mechanisms remains essential for increasing scientific understanding of the disorder and relieving the distress that it incurs.