The variation in individual clinical response to psychotropic drug treatment remains a critical problem in the management of the seriously mentally ill patient. Although a minority of patients may experience complete symptom remission, a large proportion of patients continues to experience significant psychiatric symptoms
(1), and in addition there is a subset of patients who develop drug-induced adverse events that may range from the troublesome to the life threatening
(2). Moreover, psychotropic drug efficacy may not occur until weeks after initiation of drug treatment
(3), and thus, the time period before a clinician can determine whether a specific treatment is ineffective and consider alternative pharmacotherapy can be lengthy. During this period, treated patients may experience continuous psychiatric symptoms, employment loss, social dysfunction, medical morbidity, and—in a significant proportion of patients with psychosis and affective disorders—even commit suicide
(4).
Molecular genetic approaches provide a novel method of dissecting the heterogeneity of psychotropic drug response. This field of inquiry, traditionally termed “pharmacogenetics,” provides a number of distinct advantages in the search for informative correlates of psychotropic drug response (
Appendix 1). First, an individual subject’s genotype is essentially invariable, and thus, collection of the independent measure for analysis versus treatment response can be performed at any time during treatment (or thereafter) and remain unaffected by the treatment itself
(10). Second, current molecular biological techniques provide an accurate assessment of an individual’s genotype
(11), and measurement error plays little or no role in these analyses. Third, the dramatic increase in the amount of publicly available genomic information
(12) now provides the necessary data to conduct comprehensive studies of individual genes and, perhaps, investigations of entire genomes. Finally, the ease of accessibility to genotype information by means of peripheral blood samples, coupled with advances in molecular techniques, has increased the feasibility of routine DNA collection and genotyping in large-scale clinical trials samples
(13). Therefore, pharmacogenetic approaches provide a new opportunity to identify biological predictors of psychotropic drug response, but perhaps more important, they may provide the means to determine the actual molecular substrates of psychotropic drug efficacy. In this article, we will discuss basic methods issues in executing pharmacogenetic studies, review the first generation of pharmacogenetic studies of psychotropic drug response, and consider future directions for this rapidly evolving field.
Pharmacogenetics of Antipsychotic Drugs
In psychiatry, pharmacogenetic studies have focused on three major phenotypes: clinical efficacy of antipsychotic drugs, efficacy of antidepressant medications, and development of adverse effects associated with psychotropic drug treatment.
Pharmacogenetic studies of antipsychotic drug response have focused on the atypical antipsychotic drug clozapine, perhaps because of the ease of access to blood samples from clozapine-treated patients from which to extract DNA or because clozapine’s superior efficacy in treatment-resistant populations
(28) suggests that an understanding of the genetic contributions to its effects could provide novel data on the molecular basis of antipsychotic efficacy. The initial clozapine studies were conducted either in the context of clinical trials of clozapine
(29,
30) or were based upon retrospective analyses of clozapine-treated patients
(31–
34) in ethnically heterogeneous study groups derived from Western countries, including the United Kingdom, Germany, and the United States.
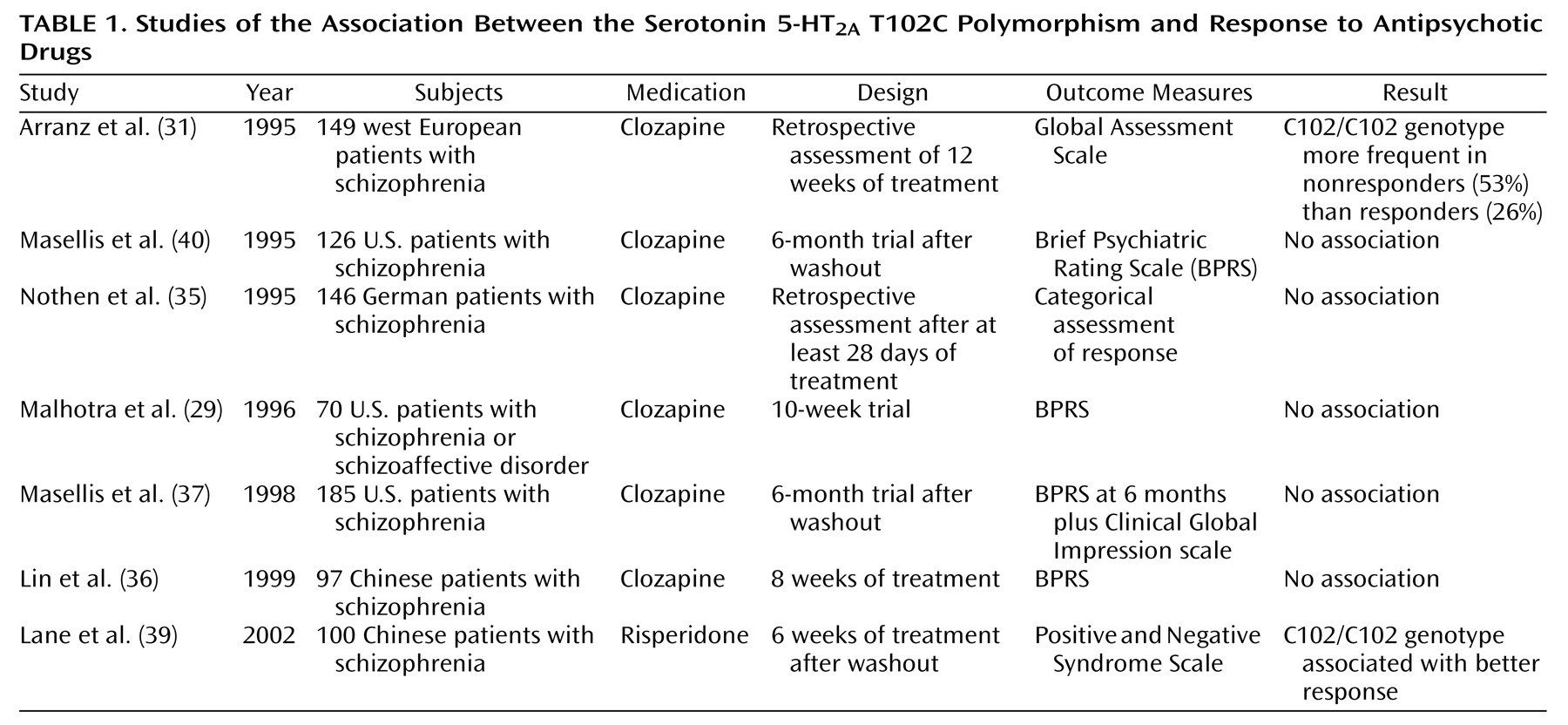
Pharmacogenetic studies of clozapine have primarily used a candidate gene approach (
Figure 1), with genetic loci in the dopamine and serotonin receptor systems—obvious candidates because of the high affinity of clozapine for these receptor subtypes. Arranz and colleagues
(31) initially attracted interest in the serotonin 5-HT
2A T102C polymorphism with a report of a significant (p=0.02) association between the 102C allele and failure to respond to clozapine in a cohort of 149 patients with chronic schizophrenia who were retrospectively assessed with the Global Assessment Scale. These data were not replicated in a series of smaller clozapine studies from independent laboratories
(29,
35–37), as well as in a study that included typical antipsychotic agents
(38) (
Table 1). 5-HT
2A T102C could be considered a relatively weak candidate polymorphism because it does not result in an amino acid substitution at the protein level and there is little evidence that it produces significant functional effects on 5-HT
2A receptor function
(40). A less common polymorphism within the 5-HT
2A gene, His452Tyr, which was not found to be in significant linkage disequilibrium (nonrandom population association) with T102C
(29), does appear to produce functional effects in vitro; however, it has not consistently been found to be associated with clozapine response
(29,
35).
Nevertheless, more recent data suggest that the positive association between 5-HT
2A T102C and clozapine response may not have been a false positive result. First, this variant is in strong linkage disequilibrium with a putatively functional polymorphism, –1438 G/A, in the promoter region of the gene
(41). Second, a meta-analysis of clozapine pharmacogenetic studies of 5-HT
2A T102C revealed an excess of the 102C allele in clozapine nonresponders in each data set, with a significant effect of this variant on clozapine response in the combined sample
(42). Since the majority of studies included in this meta-analysis were published as “negative” studies, publication bias is unlikely to have confounded these results, and the smaller studies may have provided insufficient power to detect the modest effects of this gene on a complex clinical phenotype such as clozapine response. Finally, a recent study of 5-HT
2A T102C and antipsychotic response to the atypical agent risperidone in 100 Han Chinese schizophrenia patients
(39) also identified an association, but in this instance, the association was in the opposite direction than previously observed in primarily Caucasian study groups. Definitive studies with larger group sizes, prospective clinical data, and comprehensive examination of the gene will be needed to further address the role of this gene in antipsychotic drug response.
Other serotonin-related genes that have been examined in pharmacogenetic studies of clozapine include the 5-HT
2C (30,
32,
37,
43), the 5-HT
6 (44, 45), the 5-HT
7 (45), as well as the serotonin transporter (
SLC6A4) gene
(46,
47). Although there have been some positive reports of association, there is little current evidence to suggest that variation within these genes significantly influences clozapine’s efficacy.
In addition to significant affinities for serotonin receptor subtypes, clozapine is also a dopamine receptor antagonist. Initial pharmacogenetic studies focused on the relationship between the dopamine D
4 receptor gene (
DRD4) and clozapine response
(48,
49).
DRD4 was an attractive candidate gene because of clozapine’s affinity for the D
4 receptor
(50) and the identification of a common variable number of tandem repeat polymorphisms within the putative third cytoplasmic loop of the receptor, associated with significant effects on the binding affinity of the receptor for clozapine
(51). Most groups, however, have been unable to detect a significant association between this variant and clozapine response
(48,
49,
52,
53). Ozdemir and colleagues
(54) have reported an association between a repeat polymorphism within the first intron of
DRD4 in a preliminary study of 50 patients; however, correction for multiple testing could alter the significance of these results. Clinical trials demonstrating that dopamine D
4 receptor antagonists are ineffective in the treatment of schizophrenia
(55) have also diminished enthusiasm for this receptor in antipsychotic drug efficacy.
Another obvious candidate for pharmacogenetic studies of antipsychotic drug response is the D
2 receptor gene (
DRD2). To date, all known antipsychotic drugs have potent affinities for the D
2 receptor
(56,
57), and functional brain imaging studies have suggested that D
2 receptor binding by antipsychotic agents may be “necessary and sufficient” for antipsychotic efficacy
(58). However, there are few common polymorphisms within the coding regions of
DRD2 (59), and thus, fewer studies of
DRD2 and antipsychotic drug response have been conducted, compared with the 5-HT system.
Two preliminary studies have included evidence that genetic variation within
DRD2 is associated with antipsychotic response
(60,
61). Moreover, Shäfer and colleagues
(62) examined the relationship between haloperidol, an antipsychotic drug with greater D
2 affinity than clozapine, and the
DRD2 Taq1A polymorphism and found that subjects with the A2/A2 genotype displayed poorer clinical response than heterozygous patients (no A1/A1 homozygotes were obtained). The distinction between genotypic groups was evident after 2 weeks of treatment, with 63% of the heterozygous subjects meeting response criteria versus 29% of the subjects with the A2/A2 genotype. Consistent with these results, recent preliminary data have suggested a relationship between the Taq1 A2/A2 genotype and failure to respond to risperidone
(63), another agent with greater D
2 affinity than clozapine. Finally, Suzuki and colleagues
(64) report preliminary evidence for a relationship between
DRD2 and response to the typical antipsychotic agents bromperidol and nemonapride. Therefore, many, although not all
(65), studies suggest that variation within
DRD2 may significantly influence response to antipsychotic drugs, particularly with agents with higher affinities for the D
2 receptor. It should be noted, however, that these studies used two different polymorphisms—the Taq1A variant and the promoter region variant –141C Ins/Del—that are located more than 250 kilobases (kb) apart from each other. Therefore, more comprehensive examination of
DRD2 and antipsychotic drug response may be required.
Finally, it is increasingly recognized that any individual gene’s effect on antipsychotic drug response will be modest in most populations. Therefore, examination of multiple genes and multiple single nucleotide polymorphisms (SNPs) in order to identify sensitive and specific “predictor profiles” is currently under way. A retrospective analysis of 19 candidate polymorphisms identified a grouping of six variants that provided 76.7% success in predicting clozapine response
(66). Since these data have not yet been replicated
(67), it will be critical to conduct prospective studies of this profile before these data can be considered to provide potential clinical use. Nevertheless, studies that begin to dissect the complex interactions between genetic loci and their effect on clinical phenotypes are likely to be necessary for a greater understanding of the genetic contribution to antipsychotic drug response.
Pharmacogenetics of Antidepressant Drugs
Pharmacogenetic studies of antidepressants can be classified as addressing either pharmacokinetic or pharmacodynamic effects. Genetic variants affecting the metabolism of antidepressants may change pharmacokinetic factors, such as plasma drug concentration and half-life. Polymorphisms that affect the expression or function of receptors and signal transduction molecules in the brain may alter pharmacodynamics. Both pharmacokinetic and pharmacodynamic changes can affect the efficacy and side effects of antidepressants.
Pharmacokinetic pharmacogenetic studies have focused on polymorphisms in liver cytochrome P450 isoenzymes that metabolize many antidepressant medications. The most intensively investigated gene is
CYP2D6, which encodes debrisoquine hydroxylase. Many antidepressants, including tricyclics, selective serotonin reuptake inhibitors (SSRIs), venlafaxine, and others, are metabolized primarily by debrisoquine hydroxylase
(68). The
CYP2D6 gene is highly polymorphic, with over 70 known variants (for a current classification, view the CYP P450 allele nomenclature at http://www.imm.ki.se/CYPalleles/). Homozygosity for null alleles gives rise to the poor metabolizer phenotype for debrisoquine hydroxylase characterized by no enzyme activity. Null allele heterozygosity or homozygosity for intermediate metabolic alleles gives rise to an intermediate debrisoquine hydroxylase metabolic phenotype characterized by impaired—but not absent—enzyme activity
(69–
71).
CYP2D6 gene duplications give rise to the ultrametabolic activity of debrisoquine hydroxylase
(72).
In highly controlled studies of drug metabolism performed with healthy volunteers in pharmacokinetics laboratories, the
CYP2D6 genotype has been shown to predict tricyclic and SSRI plasma concentrations
(73–
76). However, there are few studies performed in clinical settings. In a group of inpatient and outpatient geriatric patients suffering from major depression, the
CYP2D6 genotype predicted plasma nortriptyline levels at a steady state
(77). The genotype-drug concentration correlation was evident, despite the fact that, on average, the patients took 9.8 concurrent medications in addition to nortriptyline. Hence,
CYP2D6 genotyping could be useful in identifying patients likely to experience high plasma levels when treated with nortriptyline. In a retrospective study of 100 patients treated with tricyclic antidepressants and other psychotropics in a state hospital, adverse events were associated with
CYP2D6 genotypes encoding debrisoquine hydroxylase with poor metabolism
(78). These results suggest that
CYP2D6 genotypes could be useful in identifying patients likely to experience antidepressant side effects.
However, in a prospective, double-blind study of paroxetine treatment of 122 patients with geriatric major depression, Murphy et al.
(79) found that the
CYP2D6 genotype had no effect on medication-related adverse events (determined by clinician ratings), treatment discontinuations due to adverse events, and measures of treatment efficacy, such as change in Hamilton depression scale ratings over time. A number of studies have shown a relationship between the
CYP2D6 genotype (and/or the debrisoquine hydroxylase phenotype) and paroxetine levels during short- or long-term dosing
(74,
80–84), although another as yet unidentified cytochrome is also involved
(84,
85). The safety margin of SSRI agents such as paroxetine may be sufficient that impaired clearance in patients with intermediate or poor debrisoquine hydroxylase activity does not result in more adverse events. This may also be the case with other modern antidepressants that are metabolized largely by debrisoquine hydroxylase. Recommendations for dose adjustments based on
CYP2D6 genotype have been published
(86). However, until more prospective pharmacogenetic data are available, these recommendations may be premature.
In addition to metabolizing many antidepressants, debrisoquine hydroxylase can also be inhibited by these medications
(87,
88). Thus, an individual with an extensive or intermediate metabolic phenotype can be converted to a poor metabolic phenotype when treated with an SSRI such as paroxetine
(89). This could result in toxicity if the patient were concurrently taking, for example, an antihypertensive or antiarrhythmic substrate with a narrow margin of safety. Case reports have been published of fatalities after an inhibitor of debrisoquine hydroxylase was coadministered with a debrisoquine hydroxylase substrate
(90). However, despite extensive discussion in the literature on the consequences of debrisoquine hydroxylase inhibition
(76,
91), clinical data are surprisingly sparse, aside from case reports. There may be considerable interindividual variation in the degree of debrisoquine hydroxylase inhibition by agents such as SSRIs
(82,
83). Large-scale prospective studies are needed in which patients with known genotypes initiate antidepressant therapy and the effects on serum levels and side effects of concurrent medications are monitored longitudinally. In the study of Murphy et al.
(79), 26 elderly depressed subjects who were treated with paroxetine were also taking a medication classified as a debrisoquine hydroxylase substrate. These individuals had no greater severity of adverse events than other paroxetine-treated subjects, and there was no interaction between
CYP2D6 genotype, concurrent substrate medications, and adverse events. It is important to note that different results might have been obtained had the paroxetine-treated subjects concurrently taken tricyclic antidepressants, potentially toxic debrisoquine hydroxylase substrates that were not permitted in the study by Murphy et al.
(79). However, in a 4-year postmarketing survey, few clinically significant interactions between paroxetine and other medications were detected
(92). Further data are needed on this important topic, and clinicians should continue to use appropriate caution when multiple medications are prescribed.
The CYP3A4 liver enzyme is also involved in the metabolism of many psychotropics, including some antidepressants
(93–
95). A number of allelic variants at the gene encoding CYP3A4 have been identified
(92). The effects of these variants on antidepressant metabolism in clinical settings have yet to be tested.
If and when clinically useful pharmacogenetic markers for antidepressant pharmacokinetics are identified, it remains to be seen how rapidly these will be integrated into clinical practice. For example, although a strong case can be made for therapeutic monitoring of drug concentrations, there is limited clinical use of this currently available technique in making treatment decisions
(96).
A number of studies have also reported on pharmacogenetic markers that affect antidepressant pharmacodynamics. In one of the first studies involving an SSRI, Smeraldi and colleagues
(97) showed that the “short” form of a deletion/insertion polymorphism in the promoter region of the serotonin transporter gene (
SLC6A4) impaired the efficacy of fluvoxamine in a group of 53 patients with major depression (
Table 2). In some cell biology studies, the long and the short versions of the
SLC6A4 promoter polymorphism have been found to differentially affect the expression of the 5-HT transporter protein
(103,
104). SSRIs are thought to act through inhibition of the 5-HT transporter, so a genetic variant that affects the expression of this protein could affect treatment response even though the protein structure is unchanged. The same effect of the
SLC6A4 long/short promoter polymorphism was reported by Pollock and colleagues
(98) in a study of 51 geriatric patients with major depression who were treated with paroxetine. Subsequently, the Zanardi group confirmed this finding in a group of 60 paroxetine-treated depressed patients
(99). Minov and colleagues
(101) found no effect of the
SLC6A4 promoter polymorphism on response in 104 patients receiving a variety of antidepressant treatment regimens. This study is difficult to interpret because of heterogeneity in treatment regimen. However, Kim et al.
(100) studied 120 Korean patients treated with fluoxetine or paroxetine and found that the short variant of the
SLC6A4 promoter was associated with a
better treatment response. A similar result was obtained by Yoshida et al.
(102) in a study with Japanese patients. One explanation for these results may be that the effect of the
SLC6A4 promoter is dependent on ethnic background. In the studies by Kim et al.
(100) and Yoshida et al.
(102), all patients were Asian, whereas in other studies, the patients have been predominantly non-Asian. In summary, the effect of the
SLC6A4 promoter variant on SSRI treatment is the most robust antidepressant pharmacogenetic finding to date. However, contrary results from a study of Asian patients indicate that replication of this finding in other populations is necessary.
A handful of other pharmacogenetic studies aimed at antidepressant pharmacodynamics have been performed. Tryptophan hydroxylase is important in the synthesis of serotonin. Serretti and colleagues
(105,
106) studied the A218C polymorphism (located in a noncoding region of the tryptophan hydroxylase gene) and found that the A allele was associated with poor treatment response in 217 depressed patients treated with fluvoxamine and in 121 patients treated with paroxetine. Because the A218C variant does not affect the expression or structure of the tryptophan hydroxylase protein, its pharmacogenetic effect may be related to another functional polymorphism in close proximity. The 5-HT
2A receptor is located on postsynaptic neurons and may be important in antidepressant side effects
(107). Cusin and coworkers
(108) found that the C allele of the 5-HT
2A T102C SNP associated with failure to respond to clozapine was also marginally associated with poor response to SSRIs.
Most pharmacogenetic studies aimed at antidepressant pharmacodynamics have hypothesized the effects on antidepressant efficacy due to functional variation in neurotransmitter receptors and transporter proteins located within the CNS. However, many antidepressant side effects, such as those associated with the vasculature or the gastrointestinal tract, could be mediated by interactions between antidepressants and peripheral neurons and synapses. Future pharmacogenetic studies should target receptors expressed by neurons of the autonomic nervous system and on end organs as predictors of antidepressant side effects and treatment discontinuation
(109).
Although variations in genes encoding neurotransmitter receptors are a prime targets for pharmacogenetic studies, antidepressant effects on neurons involve a myriad of downstream effector molecules. Most aminergic receptors transduce signals through guanine nucleotide binding proteins (G proteins). Functional changes in these proteins could alter drug action. Zill and colleagues
(110) examined the C825T functional polymorphism in the G-protein β3 subunit gene in 76 depressed patients receiving a variety of antidepressants. TT homozygotes showed greater treatment response than did other patients, although this finding is difficult to interpret because of treatment heterogeneity. Serretti et al.
(111) found no association between D
2 and D
4 receptor polymorphisms and response to fluvoxamine or paroxetine. However, recent evidence shows that among SSRIs, only fluoxetine shows a significant effect on dopamine release
(112).
Pharmacogenetics of Mood Stabilizers
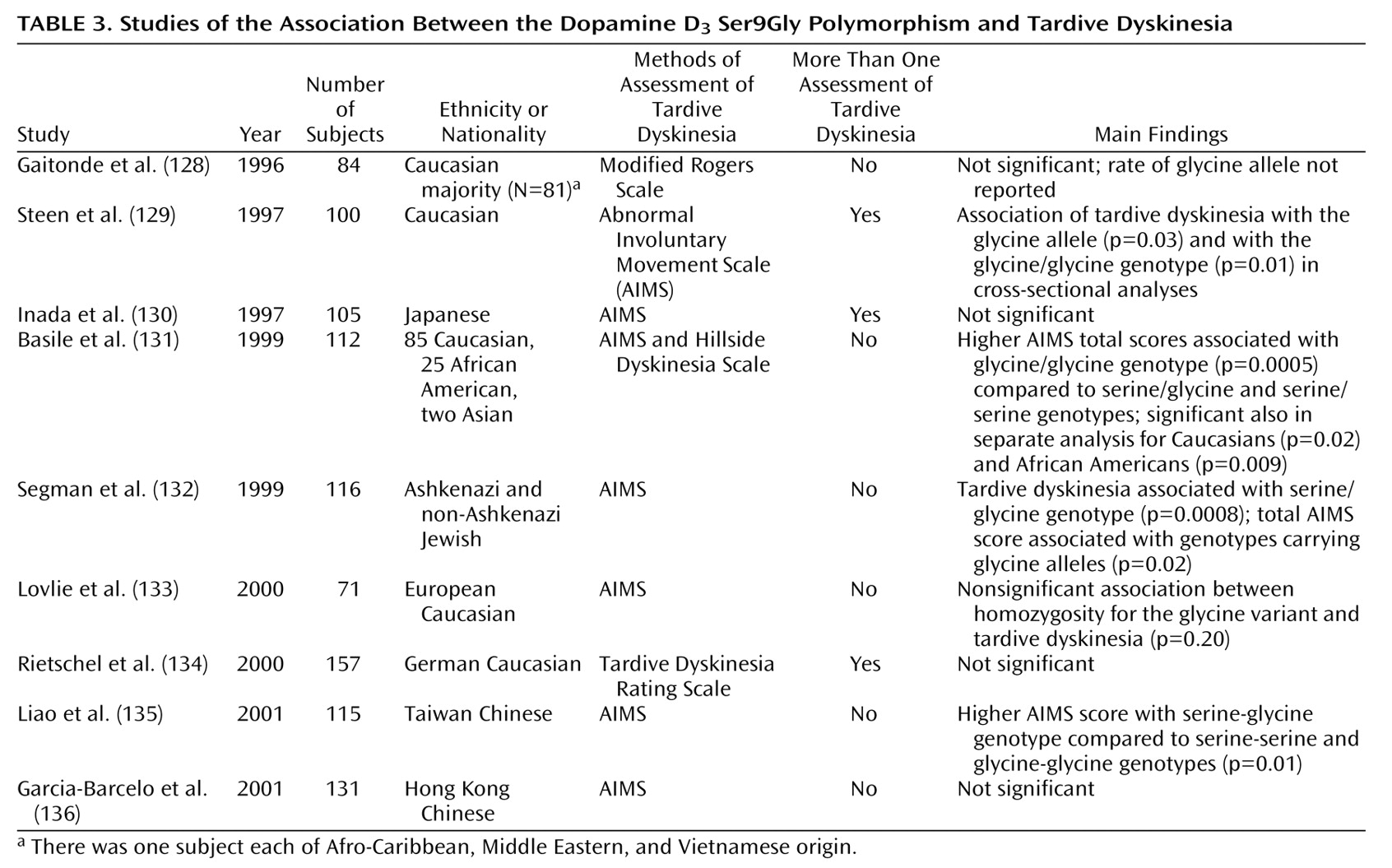
There are few pharmacogenetic studies of mood stabilizers. Selecting candidate genes for pharmacogenetic investigation is difficult because the exact mechanism of action of established mood stabilizers such as lithium remains uncertain
(113,
114). Lithium inhibits the activity of a number of enzymes, including those involved in the phosphatidylinositol cycle and phospholipase C signal transduction, although it has not been demonstrated that this action is responsible for mood stabilization. Steen and co-workers
(115) tested for associations between polymorphisms in the inositol polyphosphate 1-phosphatase gene and lithium response in two small groups of bipolar patients. In a group of 23 patients, an association between the inositol polyphosphate 1-phosphatase C973A variant was found, but in the second group of 54 patients, there was no association. Other enzymes related to inositol phosphate metabolism may be suitable targets for studies of lithium’s action, as well as unrelated enzymes that are inhibited by lithium, such as glycogen synthase kinase-3
(114).
Serretti and Smeraldi and colleagues have extensively analyzed a group of approximately 125 bipolar and depressed patients treated with lithium prophylaxis. They found no association between lithium’s efficacy and polymorphisms at the following loci: the D
2 receptor, the D
3 receptor, the D
4 receptor, the γ-aminobutyric acid (GABA) type A receptor α-1 subunit, and the 5-HT
2A and 5-HT
2C receptors
(116–
118). They found a less-than-significant association of the tryptophan hydroxylase A/A genotype with lithium response. In an expanded group of approximately 200 bipolar and depressed patients, they found no association between lithium efficacy and polymorphisms at the catechol
O-methyltransferase, the monoamine oxidase (MAO)-A, and the G-protein β3 subunit loci, but they did find a positive association with the
SLC6A4 promoter polymorphism
(119), which they previously found predicted SSRI response
(97). Finally, in a further expanded group of 443 bipolar and depressed patients, they found no association between 5-HT
2A and MAO-A polymorphisms and lithium response
(108). Interpretation of these results is difficult because of diagnostic heterogeneity in the patient groups. Furthermore, most of the candidate genes studied by these researchers were chosen for their putative association with mood disorders, rather than for a specific role in the mechanism of action of lithium.
Bipolar disorder is considered to have a high degree of heritability
(120), and it has been suggested that bipolar patients with a strong family history may respond better to lithium
(121,
122). Turecki and colleagues
(123) performed a genome-wide scan to search for markers linked with lithium response in pedigrees with bipolar disease. Linkage was found between lithium response and markers on chromosomes 15 and 7. These interesting findings will require replication in other families.