A major problem in psychiatry is how to account for the delay (about 2–6 weeks) between the initial administration of norepinephrine reuptake inhibitor antidepressant drugs and the onset of clinical antidepressant effects. In studies involving both animals and depressed patients, we and other investigators have observed a gradual increase in the release of norepinephrine and the output of its
O-methylated metabolite, normetanephrine, during long-term (e.g., starting around 3 weeks) but not short-term (e.g., less than 1 week) administration of tricyclic antidepressant drugs
(1–
4 and our unpublished data). Moreover, a gradual increase in urinary normetanephrine occurred during the period of clinical improvement (determined by a gradual and persistent decrease in Hamilton Depression Rating Scale scores) in a group of depressed patients who responded to the tricyclic antidepressant drug imipramine
(1).
The in vitro pharmacological effects of normetanephrine
(5) and the rapid in vivo metabolic inactivation of normetanephrine in the brain
(6) both suggest that normetanephrine possesses intrinsic biological activity. However, the potential pharmacological effects of normetanephrine have not been widely explored. Understanding the physiological mechanisms that contribute to this gradual increase in the release and metabolism (
O-methylation) of norepinephrine to normetanephrine during the administration of tricyclic antidepressant drugs might provide insights concerning the development of procedures to speed up the clinical antidepressant effects of these drugs
(7).
Mechanisms of Noradrenergic Transmission
We performed an exhaustive review of specific aspects of the literature on noradrenergic neurotransmission by searching the MEDLINE and Current Contents databases. “Extraneuronal uptake,” “uptake 2,” “extraneuronal monoamine transporter,” “organic cation transporter type-3,” and similar phrases were used as search terms.
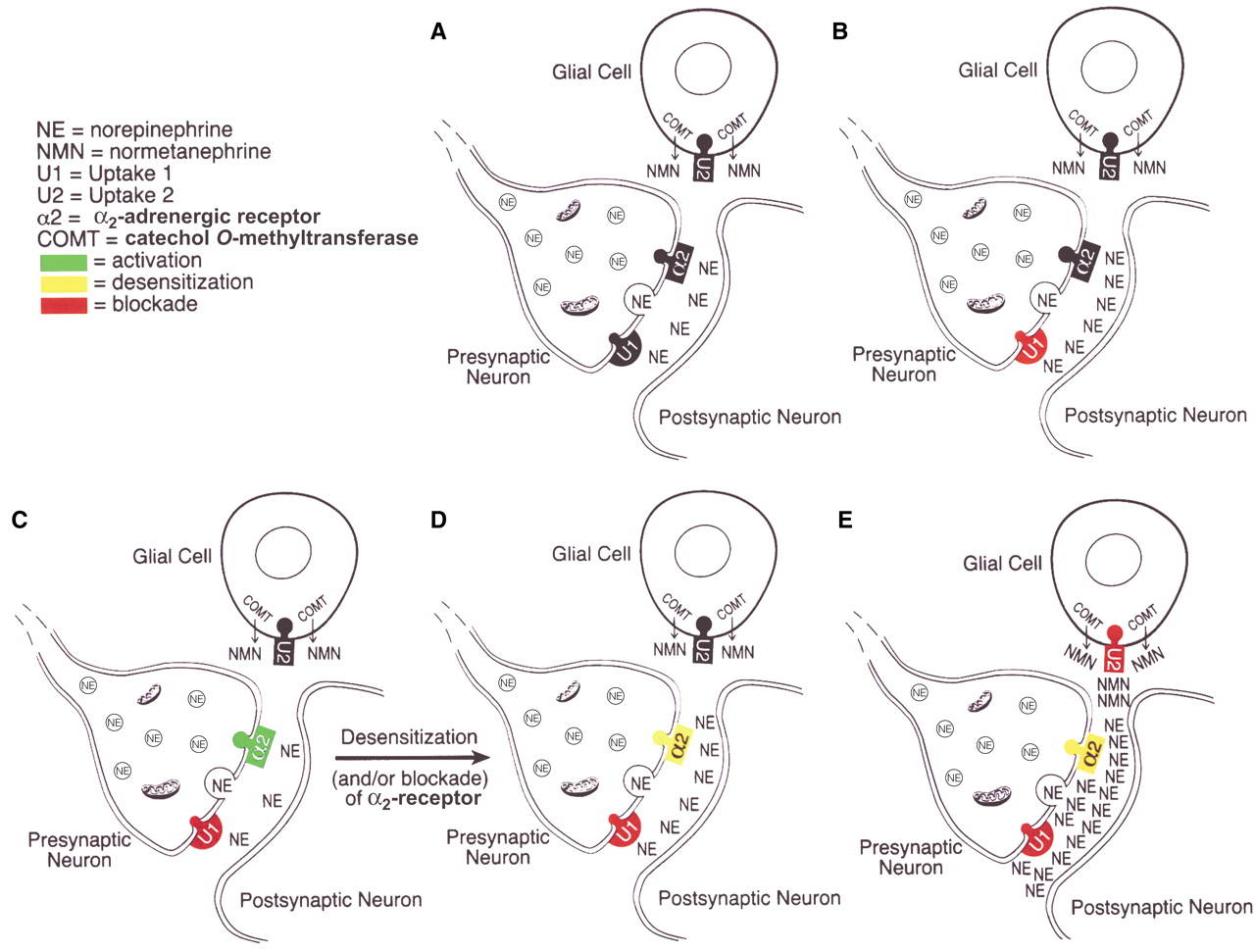
Changes in synaptic noradrenergic neurotransmission during treatment with noradrenergic reuptake inhibitors are depicted in
Figure 1. The major mechanism for the inactivation of norepinephrine that is released into the synapse is reuptake of the released norepinephrine into the presynaptic neuron of origin, i.e., so-called uptake 1, which is blocked by norepinephrine reuptake inhibitor tricyclic antidepressant drugs
(2,
8). Inhibition of presynaptic neuronal reuptake of norepinephrine (uptake 1) occurs quickly and initially produces an increase of norepinephrine at the synapse—compare part B with part A in
Figure 1. But this increase is brief because it leads to the activation of α
2-adrenergic receptors on noradrenergic nerve terminals and on cell bodies in the locus coeruleus, resulting in a decrease of locus coeruleus firing rates and a decreased release of norepinephrine from presynaptic noradrenergic neurons—compare part C with part B in
Figure 1 (9–
11).
However, during continued administration of norepinephrine reuptake inhibitor antidepressants, there is a progressive decrease in the sensitivity
(11) and/or possibly a blockade
(12) of the α
2-adrenergic receptors, leading to an increase in locus coeruleus firing rates and an increased release of norepinephrine from presynaptic neurons, thereby increasing synaptic levels of norepinephrine—compare part D with part C in
Figure 1 (3,
11)—and presumably promoting clinical antidepressant effects.
In the last several years, however, glia have been recognized to be actively involved in the process of synaptic transmission in the central nervous system
(13,
14), and norepinephrine present in the synapse or other extraneuronal spaces can also be taken up into adjacent glia by a mechanism known as uptake 2, the extraneuronal monoamine transporter
(5,
15–18). In glia, norepinephrine is converted by catechol
O-methyltransferase (COMT) to normetanephrine, which is a potent inhibitor of uptake 2
(5,
19). Thus, the formation of normetanephrine following the uptake of norepinephrine by the extraneuronal monoamine transporter, uptake 2, inhibits further uptake of norepinephrine by uptake 2—compare part E with part D in
Figure 1—producing a marked increase in levels of norepinephrine in the synapse.
Findings
In a recent study of depressed patients treated with desipramine, the sum of urinary levels of norepinephrine plus normetanephrine, expressed in micrograms per 24 hours, at baseline (mean=278, SD=90) and after 1 week (mean=241, SD=79), 4 weeks (mean=333, SD=97), and 6 weeks (mean=353, SD=125) of treatment was significantly increased over time according to a random effects regression analysis (J. Hennen, personal communication). Compared to baseline values, this sum was slightly decreased after 1 week of treatment but was significantly increased (p<0.01, postmodeling two-tailed t test) after both 4 and 6 weeks of treatment with desipramine.
Moreover, we observed that, when compared to baseline values, the ratio of urinary norepinephrine to normetanephrine was significantly increased (p<0.001, postmodeling two-tailed t test) after 4 and 6 weeks of treatment with desipramine but not after 1 week of treatment (our unpublished data).
These findings are compatible with the hypothesis that normetanephrine is indeed blocking uptake 2, thereby decreasing the access of norepinephrine to COMT and, thus, the inactivation of norepinephrine by enzymatic conversion to normetanephrine—compare part E with part D in
Figure 1.