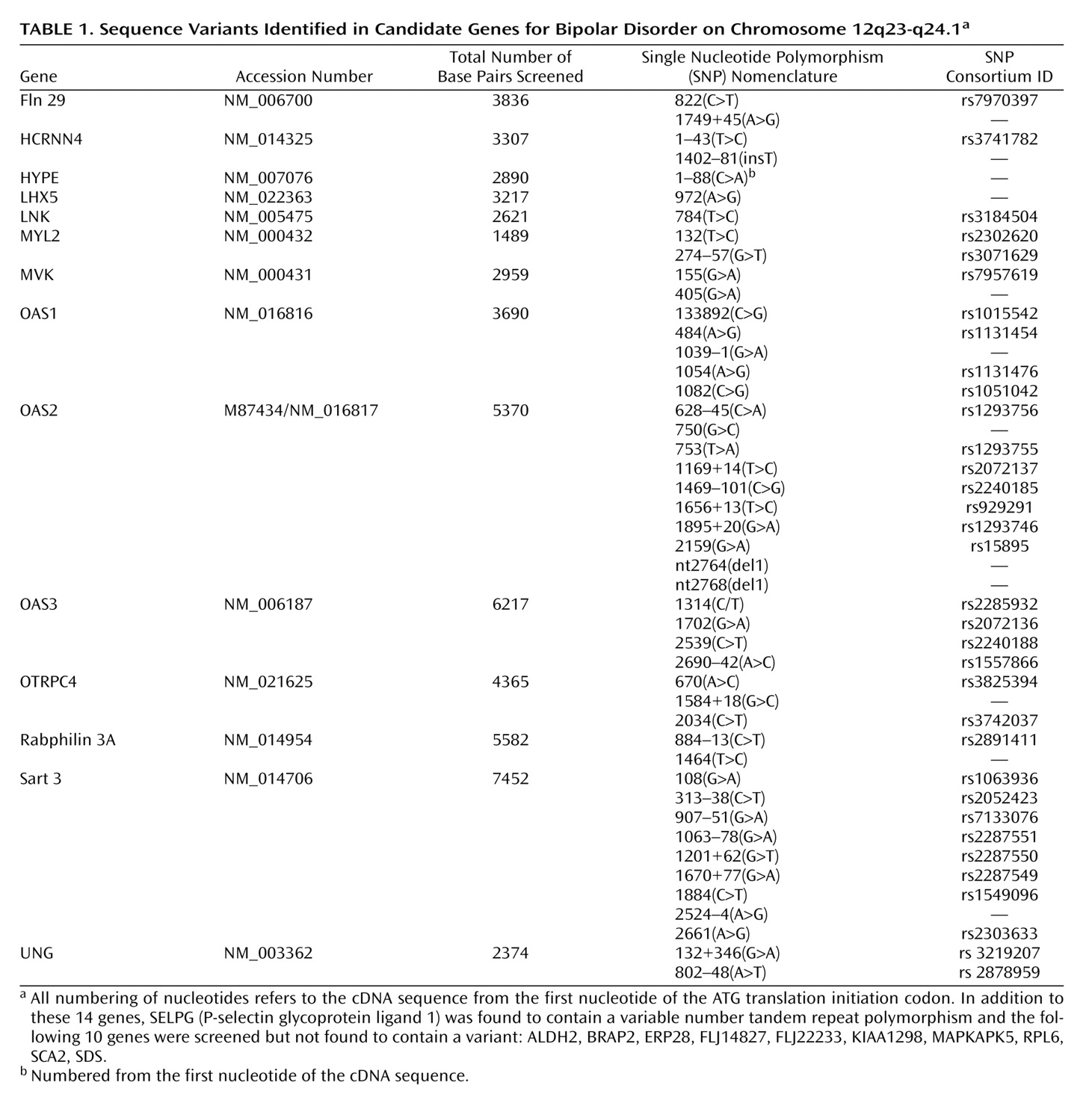
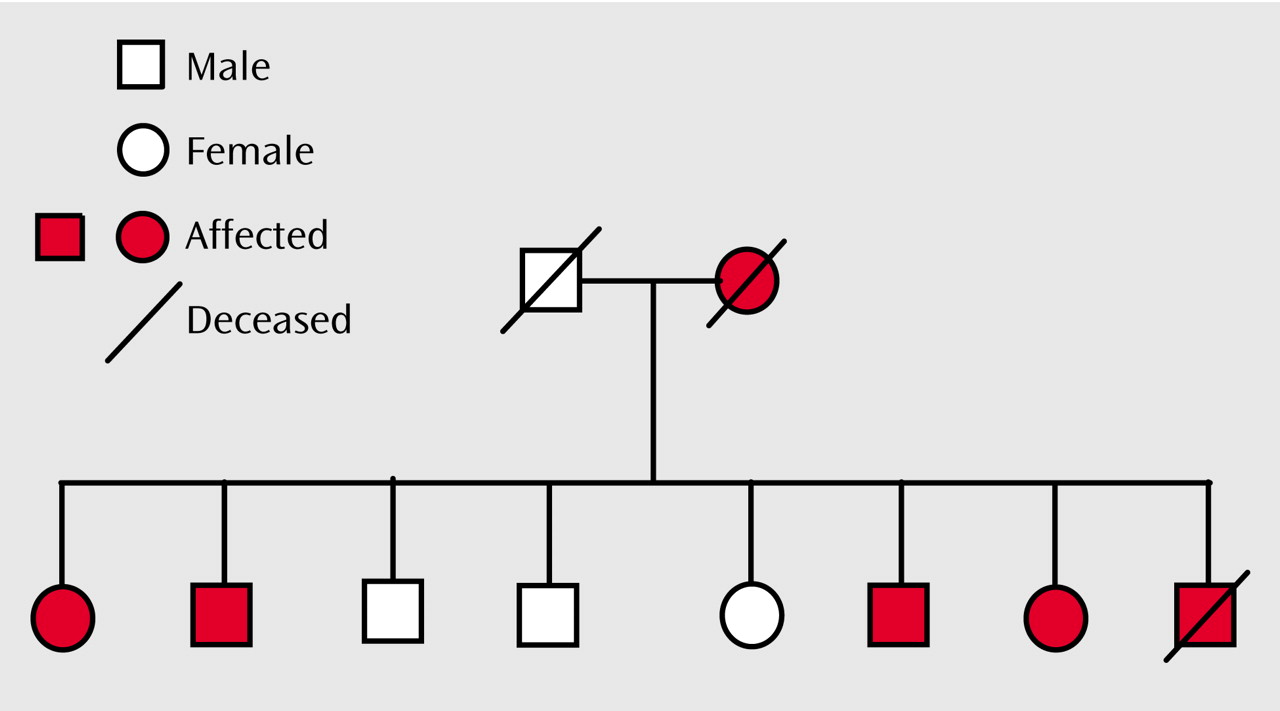
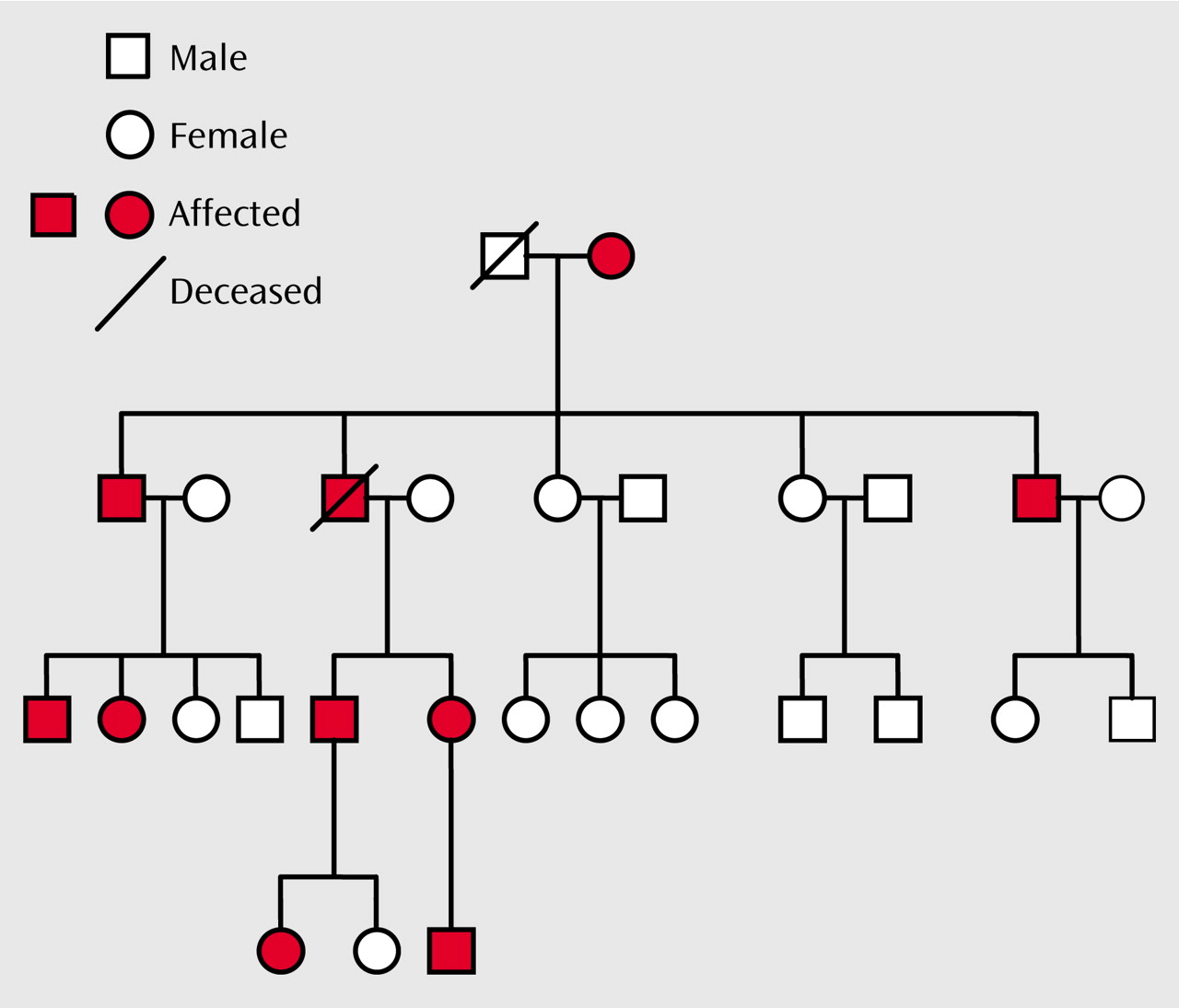
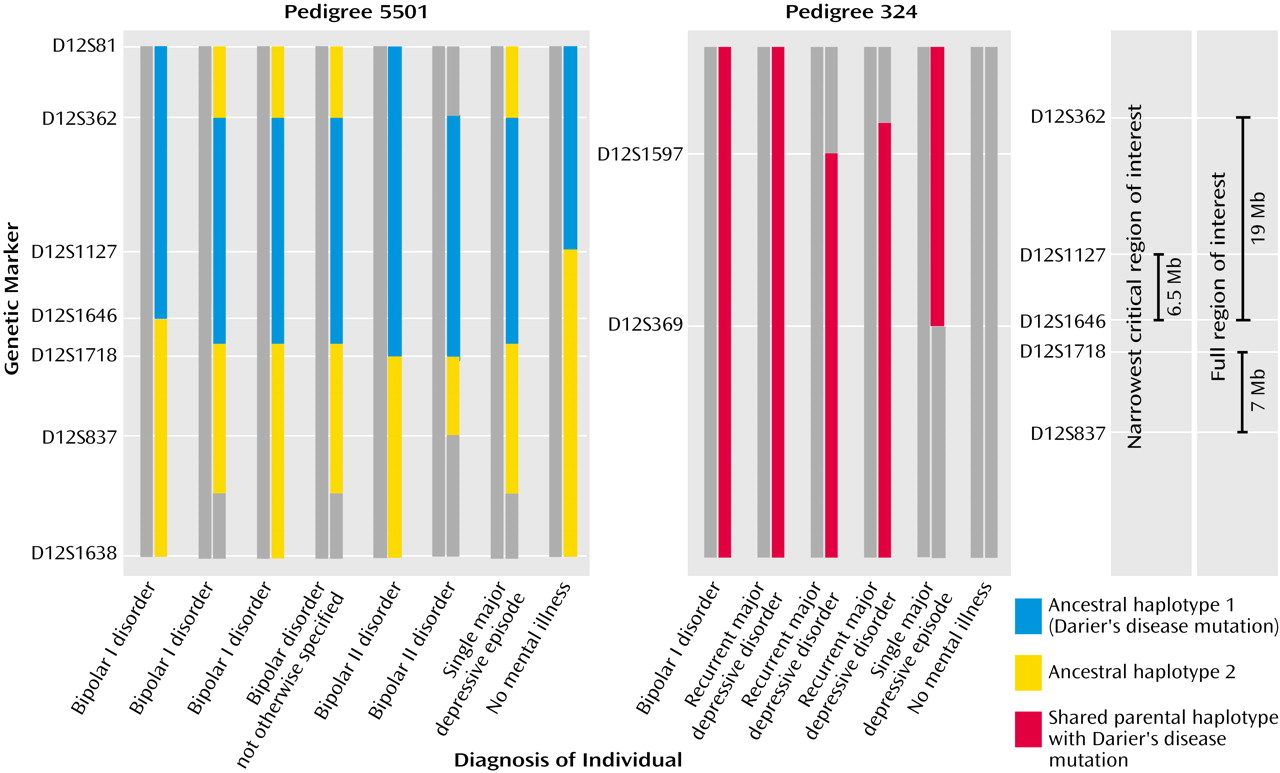
There is convincing evidence from family, twin, and adoption studies for the importance of genes in determining susceptibility to bipolar disorder
(1–
3). The relative recurrence risk (compared with population risk) for narrowly defined illness (DSM-IV bipolar I disorder) in siblings of a proband, λ
S, is approximately 8. The monozygotic probandwise concordance rate is approximately 60% (λ
MZ=60), compared with a dizygotic recurrence risk that is similar to that for siblings (λ
DZ=8)
(4,
5). Within the families of bipolar probands there is a higher than average rate of unipolar depressive disorders
(6), indicating that bipolar susceptibility genes can be expressed in a broad spectrum of mood phenotypes. To explain the inheritance of bipolar disorder within the general population, it is necessary to invoke interacting (epistatic) oligo-genes or more complex genetic models
(7). However, there have been relatively rare descriptions of families in which inheritance is consistent with the segregation of alleles of major effect (e.g., references
8–11). We have previously described two such families in which there is cosegregation of Darier’s disease (a rare, autosomal dominant skin disease) and major affective disorder: pedigree 324
(12) and pedigree 5501
(13). The Darier’s disease gene was mapped to the 12q23-q24.1 region
(14,
15). The cosegregation in pedigrees 324 and 5501 is consistent with genetic linkage between the Darier’s disease gene and a gene that, in these families, confers risk for major mood disorders according to a highly penetrant, autosomal dominant mode of inheritance. The presence of a susceptibility gene for mood disorders in the 12q23-q24 region is supported by linkage studies of bipolar disorder
(11,
16–21) and of recurrent major depression
(22,
23), an observation consistent with the hypothesis that at least some susceptibility genes contribute to both disorders. As is often the case for complex genetic disorders, the linkage signals span a region of several tens of centimorgans.
The locus causing Darier’s disease has been identified as ATP2A2, which encodes a sarcoplasmic/endoplasmic reticulum calcium-ATPase, SERCA2
(24). One hypothesis that deserves consideration is that in these two pedigrees bipolar disorder could be due to pleiotropic effects of mutations in ATP2A2
(12). This possibility finds some support from our observation of three individuals with both Darier’s disease and bipolar-spectrum mood disorder who had mutations in the same domain of ATP2A2
(25). However, against this are 1) a lack of evidence demonstrating a population-level association between bipolar disorder and Darier’s disease, 2) incomplete cosegregation of bipolar disorder and Darier’s disease in pedigree 5501
(13), demonstrating that the Darier mutation in this family is not on its own sufficient to cause bipolar disorder, and 3) our failure to identify ATP2A2 mutations in bipolar individuals from families showing greater allele sharing at this chromosomal region
(26). Thus, the current data suggest that the observed cosegregation is the result of linkage rather than pleiotropy. This is the hypothesis underpinning the current study, in which we 1) delineated the 12q23-q24 region most likely to harbor a bipolar susceptibility gene and 2) systematically studied 25 known, annotated genes within the narrowest region of interest in order to search for mutations that confer risk of bipolar disorder according to a highly penetrant autosomal dominant model in these families.
Discussion
We undertook molecular genetic studies of two pedigrees in which Darier’s disease and major affective disorder cosegregate, in order to delineate the region of chromosome 12q23-q24 most likely to contain a susceptibility gene for bipolar spectrum illness. In each pedigree the pattern of inheritance of mood disorder was consistent with a major gene model. This is certainly not the usual pattern of inheritance of bipolar disorder
(35), and therefore it is important to clarify explicitly the assumptions that underpin the approach we took. We assumed that in each pedigree, a variant exists that acts as a highly penetrant autosomal dominant mutation and that the pathogenic variant(s) (which might be different in the two pedigrees) is rare because otherwise such pedigrees would be common in the population.
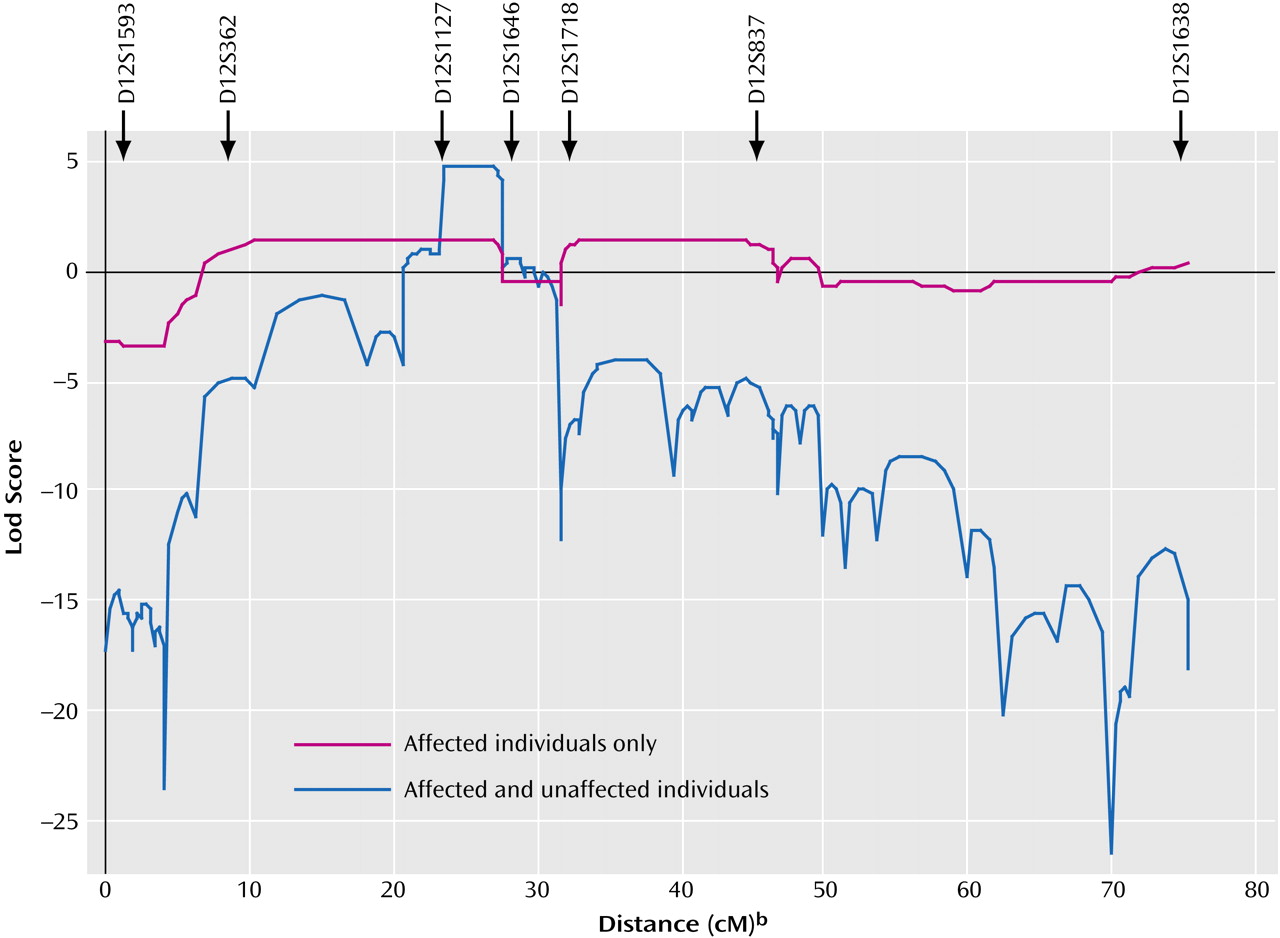
If we assume that all individuals with any mood disorder in each family carry the susceptibility variant and that those without such a diagnosis do not (in formal genetic terms, this is a completely penetrant autosomal dominant model without phenocopies), then our linkage and haplotype analyses are consistent in identifying a narrowest region of interest of 5 cM (6.5 Mb) (
Figure 3 and
Figure 4). Alternatively, if we focus on the most clear-cut bipolar cases (i.e., core bipolar phenotype) and exclude unaffected subjects from the analyses to allow for reduced penetrance, our analyses are consistent in identifying a broader “full region of interest” that spans 36 cM (26 Mb) (
Figure 3 and
Figure 4). The narrowest region of interest can be considered as our
most likely location for harboring the susceptibility variant operating in these pedigrees and, therefore, was the priority segment in which we focused the intensive direct gene analysis. The full region defines the boundaries
outside of which it is very unlikely that a susceptibility variant of major effect operates in these pedigrees.
As in all genetic studies, our power to refine the location of susceptibility loci is limited by the number of subjects (i.e., the number of informative meioses within our two pedigrees). Improved localization could be achieved if other, preferably large, families demonstrating linkage in this region can be identified and studied with similar approaches.
As mentioned in the introduction, several other groups have reported evidence supporting linkage of bipolar disorder susceptibility to chromosome 12q23-q24. Which of these findings are compatible with our regions of interest, and what is added by our data?
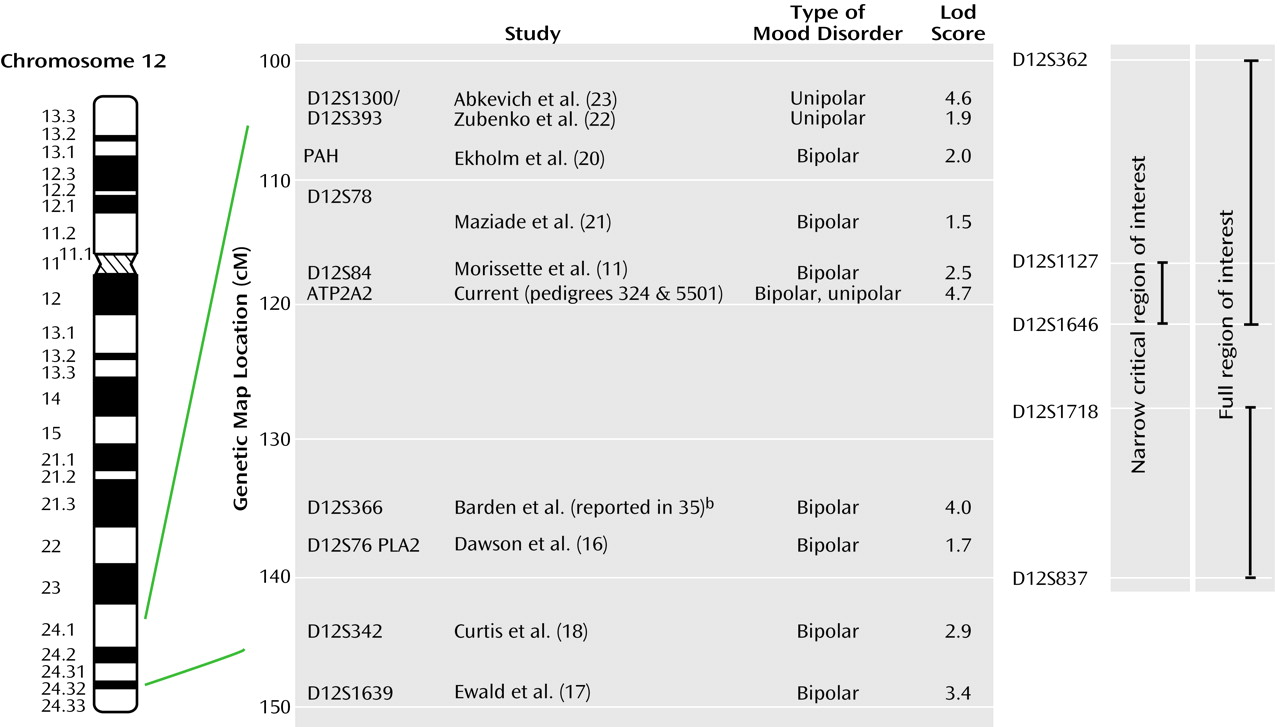
Figure 5 shows schematically our findings and the other positive reports across this region. It is well known that in complex diseases the localization of susceptibility genes is poor, with signals often being tens of centimorgans away from the true locus and regions of positive linkage being broad
(36). On this basis, the localization provided by haplotype analysis in our pedigrees consolidates the linkage findings reported by Dawson et al.
(16), Ekholm et al.
(20), Morissette and colleagues
(11), Ewald et al.
(9), Curtis et al.
(18), and Maziade et al.
(21), which all yield evidence either inside or within a few centimorgans of our region. Further, the recent genome scan meta-analysis for bipolar disorder that used the ranking method identified the 12q23 region (i.e., the proximal part of our region of interest) as one of 23 “bins” (out of a total of 120) that achieved nominal statistical significance under at least one model
(37). Indeed, our data together with these other findings strongly suggest the presence of at least one susceptibility gene for bipolar disorder within the 26-Mb full region of interest. However, the telomeric localization by linkage disequilibrium reported by Degn et al.
(19) lies outside our core phenotype region, raising the possibility that an additional mood disorder susceptibility gene resides within the distal 12q24 region. This possibility may represent the human counterpart of the observation in animal studies that multiple quantitative trait loci may be responsible for linkage peaks in genome scans (e.g., reference
38). It is of particular interest that the linkage signals in the recent genome scans for unipolar depression
(22,
23) overlap with, and lie at the
centromeric end of, our full region of interest. This suggests that genetic variation in this region may influence susceptibility to both bipolar and unipolar forms of major affective disorder. Our own pedigrees are fully consistent with this possibility in that both include unipolar as well as bipolar cases.
We focused our direct gene analysis on the narrowest region of interest. Within the context of our hypotheses and methods, we can exclude the sequences we examined from harboring a major susceptibility mutation in the two pedigrees examined. There are a number of caveats with regard to these results. First, we examined only the most well-characterized genes in the narrowest region of interest. These data will inform the search by other research groups, but it is important to recognize that additional sequences in the region remain to be studied. Second, we need to consider the adequacy of our mutation detection method. The genes were screened by denaturing high-performance liquid chromatography, which has been proven to be a sensitive method of mutation detection with nearly 100% detection efficiency when used with both the recommended DNA strand melting temperature (RTm) and RTm+2°C
(32,
39). We can, thus, be confident that we are unlikely to have failed to detect heterozygous variants within the DNA sequence investigated. Third, we did not systematically examine regions that may regulate gene expression. Our rationale was that with the existing state of annotation of the genome, it is impossible to systematically identify all important regulatory elements, and at any rate, mutations in exons dominate the mutation profile of alleles of major effect. Fourth, these genes have been excluded only under our initial model hypothesizing a highly penetrant dominant mode of inheritance. It is possible that there is a more complex mode of inheritance contributing to bipolar disorder within these pedigrees, and indeed, that is the rationale for using a more relaxed genetic model to define the broader, full region of interest. Exclusion of variants under such a model requires a different strategy involving systematic examination of large groups of cases and comparison subjects to seek either linkage disequilibrium, in the case of common disease variants, or multiple rare disease-associated variants. Therefore, while we are continuing to investigate predicted and partially identified genes within the narrowest region of interest according to the highly penetrant dominant model, as a complementary strategy we are undertaking systematic linkage disequilibrium analysis in outbred study groups, combined with direct analysis of specific candidate genes of high prior interest.