Drug addiction has been conceptualized as a chronic relapsing disorder characterized by compulsive drug-taking behavior with impairment in social and occupational functioning. From a psychiatric perspective, drug addiction has aspects of both impulse control disorders and compulsive disorders
(1) . Impulse control disorders are characterized by an increasing sense of tension or arousal before the commission of an impulsive act; pleasure, gratification, or relief at the time of commission of the act; and following the act, there may or may not be regret, self-reproach, or guilt
(2) . In contrast, compulsive disorders are characterized by anxiety and stress before the commission of a compulsive repetitive behavior and relief from the stress by performing the compulsive behavior. As an individual moves from an impulsive disorder to a compulsive disorder, there is a shift from positive reinforcement driving the motivated behavior to negative reinforcement driving the motivated behavior. Drug addiction has been conceptualized as a disorder that progresses from impulsivity to compulsivity in a collapsed cycle of addiction composed of three stages: preoccupation/anticipation, binge/intoxication, and withdrawal/negative affect
(3) . Different theoretical perspectives ranging from experimental psychology, social psychology, and neurobiology can be superimposed on these three stages, which are conceptualized as feeding into each other, becoming more intense, and ultimately leading to the pathological state known as addiction
(3) . The thesis of the present review is that excessive drug taking in dependent animals can be studied in animal models, involves important perturbations in the stress response systems of the body, and contributes to both the positive reinforcement associated with impulsivity (binge stage of the addiction cycle) and the negative reinforcement of the withdrawal/negative affect stage of the addiction cycle.
For several years, the desirability of developing rodent self-administration models that more closely mimic the human patterns of self-administration of specific drugs of abuse has been a focus of research by both the Koob and Kreek groups. The most frequently used models are appropriate for assessing the impact of very modest exposure and first exposure to a drug of abuse as well as continued exposure to very modest amounts of a drug of abuse on a limited basis but are not designed to study addiction-like patterns of self-administration. From a clinical standpoint, it has long been recognized and well established that both opiate addicts and cocaine addicts have very different patterns of self-administration. For opiate abusers (primarily heroin abusers), intermittent opiate use is the initial pattern of intake and may continue for unpredictable lengths of time, ranging from 1 to 2 weeks up to several years or even a lifetime (for instance “weekend chippers”). On the other hand, it has been quite well documented from the very earliest work on developing an agonist treatment modality at Rockefeller University that heroin addicts (and other short-acting opiate addicts) self-administer their drug of abuse daily and at multiple times during the day at evenly spaced intervals
(4) . These intervals are well-planned, either to prevent the onset and development of withdrawal symptoms or to maximize the limited euphorigenic effects that may be forthcoming from any single dose of a short-acting opiate, such as heroin, especially as tolerance develops. However, the heroin addict at the end of the day does eventually go to sleep for overnight rest. After awakening in the morning or midday, signs and symptoms of withdrawal have appeared, and thus acquisition of the “morning dose” of heroin immediately occurs. For cocaine addicts, the most common mode of self-administration after initial use is a binge pattern, in which from three to a dozen or more self-administrations of cocaine will occur at 30-minute to 2-hour intervals in a volley, or binge, with no cocaine self-administered for 1 day or even 1 week after a long string of binge self-administrations.
In the present review, we will build on earlier work
(1,
5 –
27) to explore the role of the brain and hormonal stress systems in addiction. To accomplish this goal, we will primarily explore extension of previously used self-administration models to include animal models of the transition to addiction, such as 1) extended access to drug self-administration, 2) long-term exposure before self-administration, 3) and the use of very high doses per unit self-administered (compared to more conventional moderate and low doses).
New Findings Have Not Negated Our Earlier Hypotheses but Have Reinforced Them
In addition to the major sources of reinforcement in drug dependence, both the persistence of ongoing addiction and relapse to drug addiction days, months, or years after the last use of the drug may be due, in part, not only to conditioned positive and negative reinforcement but also to the negative reinforcement of protracted abstinence when it exists (as, for instance, has been well documented in the case of opiate addiction) and also to much more subtle factors that result from long-term changes or abnormalities in the brain after long-term exposure to a drug of abuse due to intrinsic neuroplasticity of the brain
(4,
28,
29) . These changes may contribute to a general, ill-defined feeling of dysphoria, anxiety, or abnormality and also could be considered a form of protracted abstinence
(3) . In addition, genetic factors and early environmental factors may contribute to variations or abnormalities in neurobiologic function that may render some individuals more vulnerable, both to acquisition of drug addiction and relapse to drug use after achieving the abstinent state
(5) .
What also is new since 1998 is substantial evidence for our subhypothesis that corticotropin-releasing factor (CRF), through its actions in activating the hypothalamic-pituitary-adrenal (HPA) axis and brain stress systems in the extended amygdala, is a key element contributing to the emotional dysregulation of drug dependence.
Animal Models of Excessive Drug Taking in Dependent Animals
Most animal self-administration models to date have sessions for only 1 or 2 hour per day with no access for longer periods of time, such as 6 to 24 hours, which would more closely mimic the human condition. Also, the doses per injection allowed in animals are usually low to extremely low, with the presumed scientific purpose of minimally altering neurobiological systems to elucidate threshold effects with the practical reason of preventing accidental animal overdose. Neither of these constraints pertain to humans; heroin addicts administer maximal doses monetarily affordable and within the limits of physiologic tolerance to prevent accidental opiate overdose (although this sometimes does occur on the street with surges in purity of heroin). Cocaine addicts similarly self-administer cocaine to the extent of funds available at the time within the limits of tolerable side effects, primarily jitteriness, nervousness, dysphoria, and depression.
The Koob and Kreek groups have created new models that more closely mimic the human condition. In the Koob group, extended-access models have been developed for long-term self-exposure, extinction, reexposure, and relapse and have included very long-term studies for each of several drugs of abuse
(30 –
33) . In the Kreek group, even longer sessions of extended access have been used for short-term through long-term exposure, and some studies have involved acquisition, extinction, and rechallenge
(34 –
38) . High and moderate doses of cocaine and morphine have been used in addition to the more usual low and very low doses per injection. Extended access to drugs of abuse produces dramatic increases in drug intake over time that mirror the human condition and that at a neurobiological level more clearly mimic the investigator-administered binge pattern.
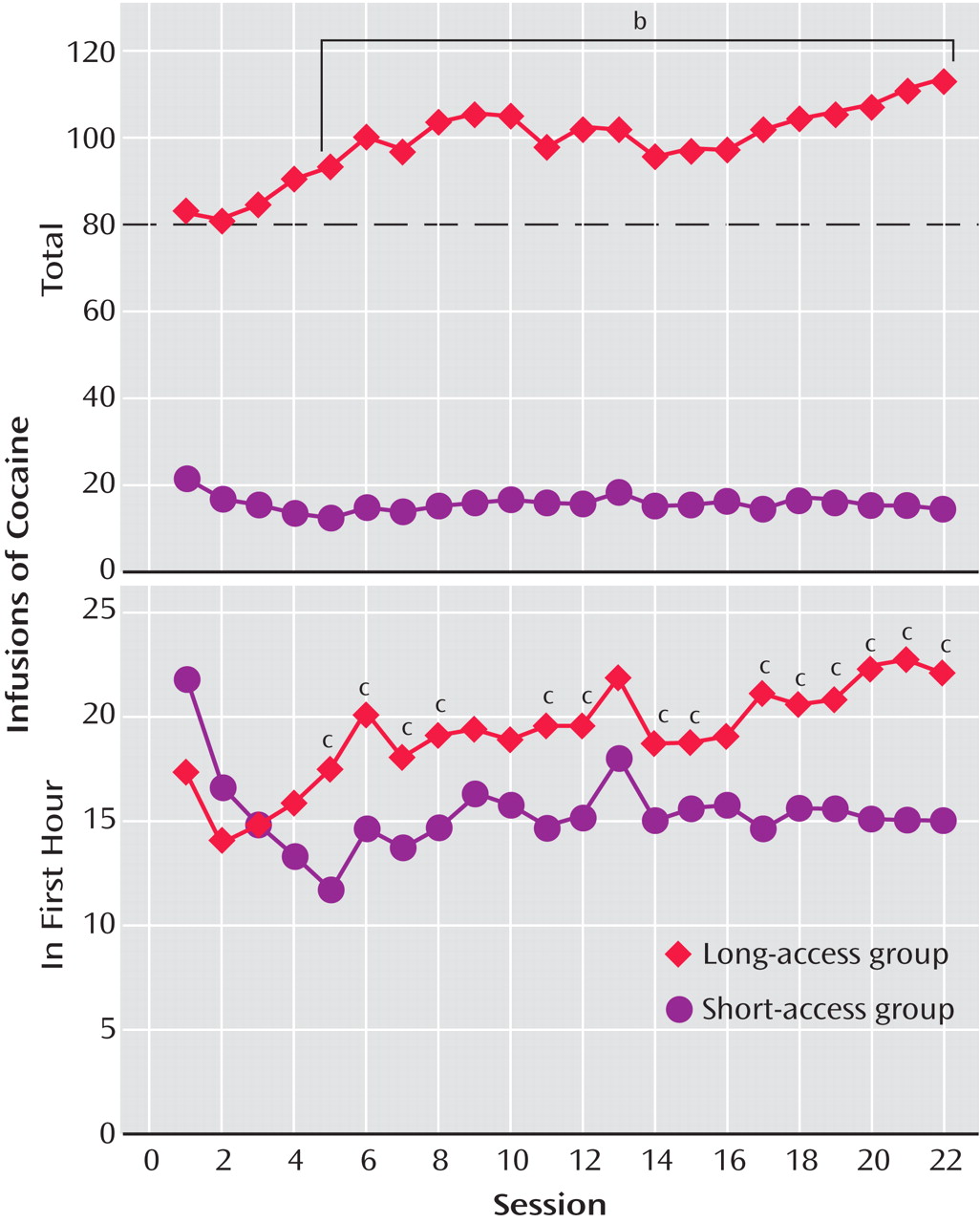
To explore the possibility that differential access to intravenous cocaine self-administration in rats may produce different patterns of drug intake (the Koob group), rats were allowed access to intravenous self-administration of cocaine for 1 hour and 6 hours per day
(30,
34 –
36,
38,
39) (
Table 1 ). With 1 hour of access (short access) to cocaine per session through intravenous self-administration, drug intake remained low and stable, not changing from day to day as observed previously. In contrast, with 6-hour access (long access) to cocaine, drug intake gradually escalated over days
(30) (
Figure 1 ). In the escalation group, there was increased early intake, sustained intake over the session, and an upward shift in the dose-effect function, suggesting an increase in the hedonic set point.
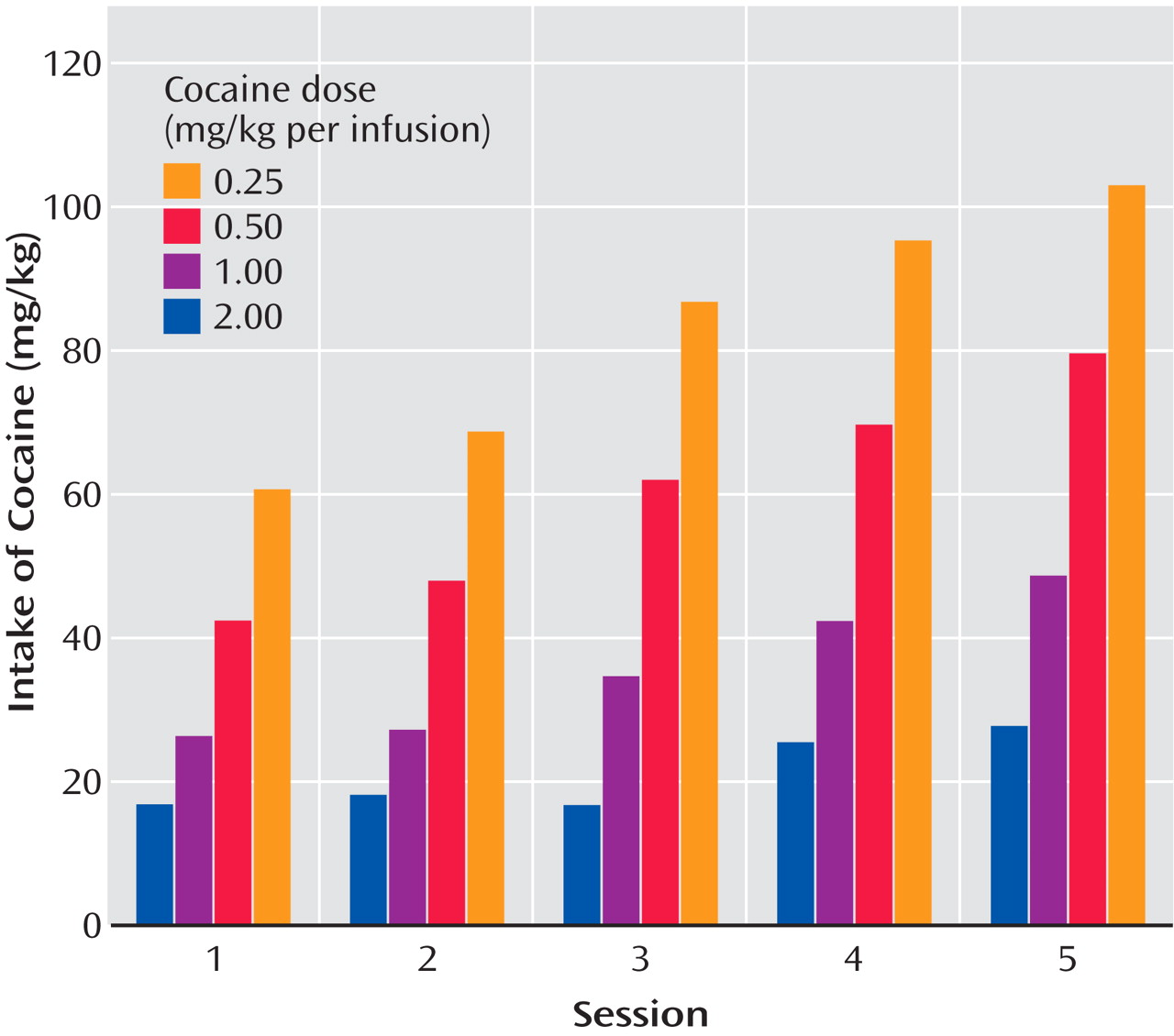
In a similar 10-hour extended-access model (the Kreek group), intravenous cocaine was self-administered by randomly assigned rats allowed to self-administer 0.25, 0.50, 1.00, or 2.00 mg/kg per infusion intravenously in a continuous schedule of cocaine reinforcement during five consecutive daily 10-hour sessions
(34) (
Table 1 ). When data from the animals self-administering any dose of cocaine were collapsed as a single group, the mean amount of self-administered cocaine exceeded 60 mg/kg per day, significantly greater than in our investigator-administered binge administration with 3×15 mg/kg cocaine per day (for a total of 45 mg/kg per day). In addition, when data from animals were analyzed by their randomly assigned group, the total daily dose administered by animals allowed to self-administer the highest and intermediate doses of cocaine (2.00 and 1.00 mg/kg, respectively) had a much steeper incremental daily total amount of cocaine self-administered than did low and very low dose groups. Animals allowed access to the highest (2.00 mg/kg per injection) dose were administering over 100 mg/kg by the end of the 5-day period. The slope of this acquisition was much steeper in the moderate and high-dose groups than in the animals allowed to self-administer very low (0.25 mg/kg) or low (0.50 mg/kg) doses of cocaine
(34,
35) (
Figure 2 ).
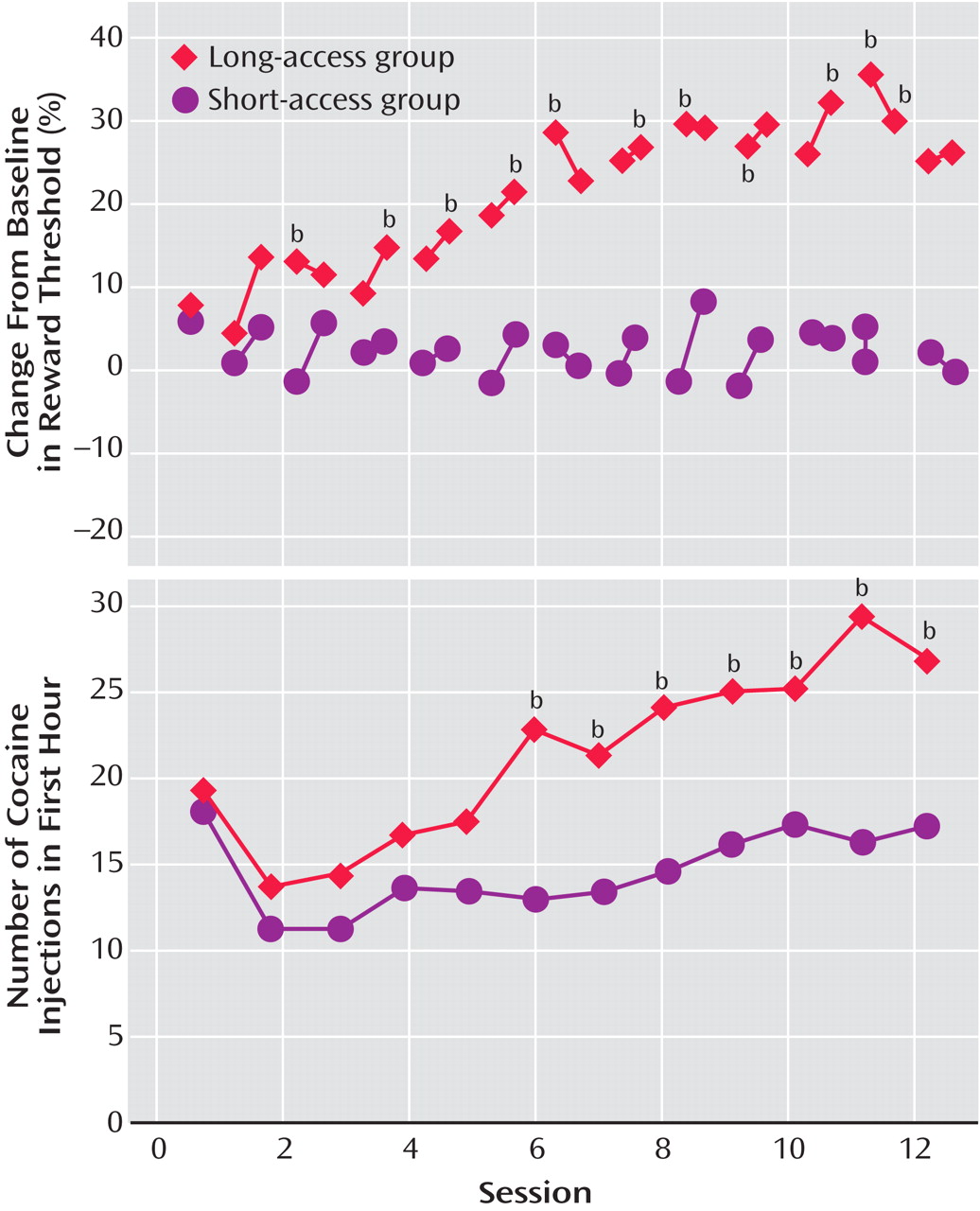
In further studies of the escalation in drug intake with extended access, rats were randomly assigned to short-access and long-access groups
(36) (
Table 1 ). The short-access animals were tested daily for multidose self-administration for 3 hours. The long-access animals were tested initially with multidose self-administration over 3 hours. Over the next 7 hours, the animals were allowed to self-administer a relatively high dose of cocaine (2.0 mg/kg). After 14 days, lever pressing was extinguished in 10 consecutive 3.5-hour extinction sessions. Following extinction, the ability of a single noncontingent investigator-administered infusion of cocaine at 0, 0.50, or 2.00 mg/kg to reinstate extinguished lever pressing was studied. Self-administration was not altered over time in the short-access rats. However, a general escalation of cocaine intake was found in the long-access, high-dose rats, which showed an increased susceptibility to reinstatement.
Similar changes in the self-administration of heroin and alcohol have been observed in animals with more prolonged access
(31) or a history of dependence
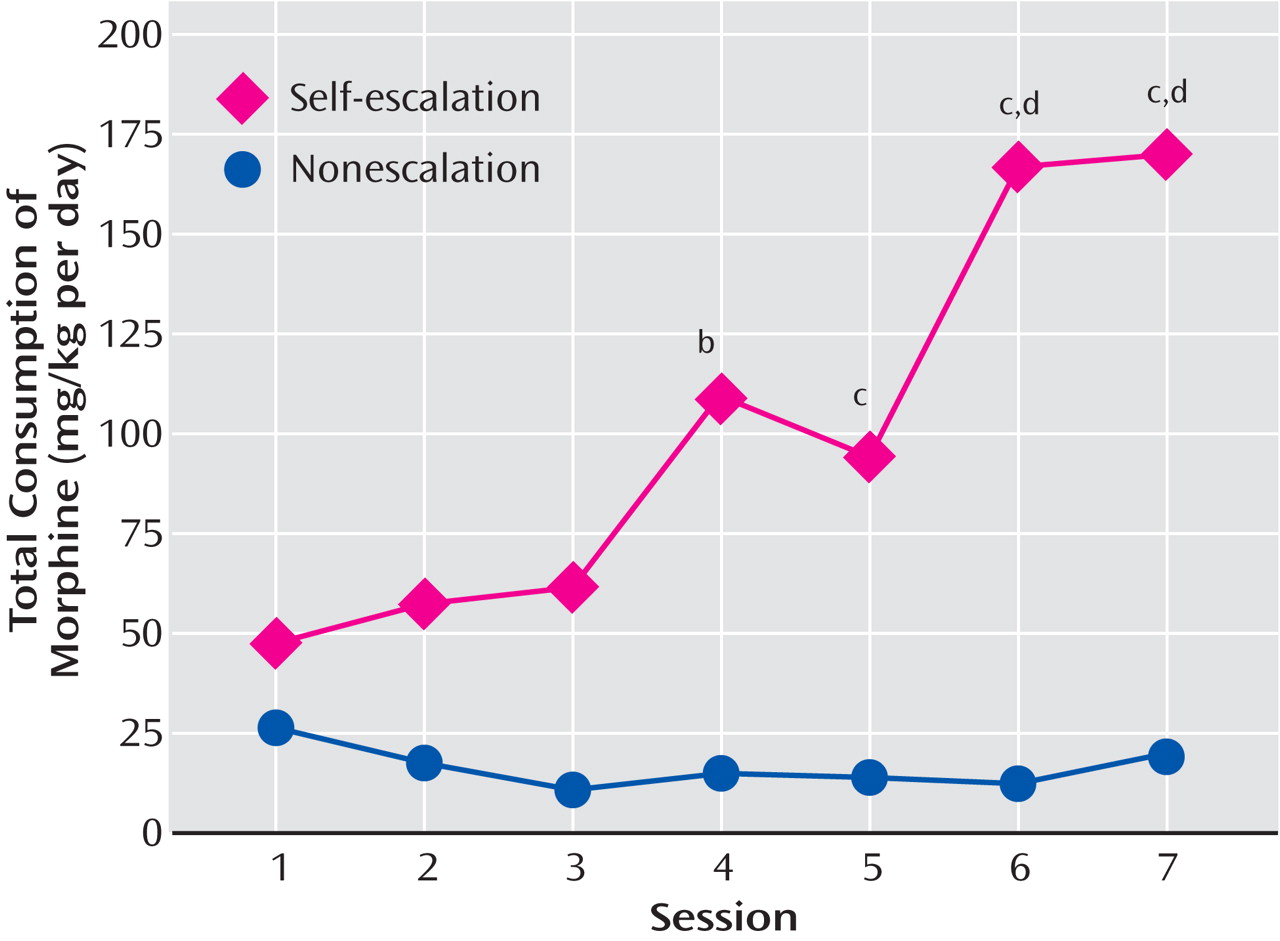
(39) . In related work, prolonged access to escalating doses of morphine in a rat self-administration model in which the animals self-regulated the dose of drug showed that repeated intake of opioids is associated with significant escalation in intake. Rats self-administered one of three doses of morphine (0.30, 1.00, or 3.00 mg/kg per infusion) during 7 daily 4-hour (short-access) sessions. In a second experiment, all animals were allowed 18-hour sessions of self-administration for 7 consecutive days and were randomly assigned to a self-escalation, individual-choice group or a fixed morphine dose group
(38) (
Table 1 ). For the short-access 4-hour sessions, the dose of 0.30 mg/kg morphine per injection did not adequately support stable self-administration, but higher doses did. The animals who had 18-hour extended access in the self-escalation model even on day 1 administered more morphine than the fixed-dose group. The total daily consumption from day 1 was approximately 45 mg/kg and with escalation reached significance by day 4 and continued through day 7. By day 7, the animals were self-administering an average of 165 mg/kg per day of morphine. These results dramatically demonstrate escalation in morphine intake, consistent with studies of escalation in heroin intake described by heroin addicts
(38) (
Figure 3 ). Similar results were obtained with 23-hour access to heroin, in which rats reached daily levels of up to 3.0 mg/kg per day and showed significant changes in circadian patterns that paralleled the escalation in intake
(40) .
Ethanol-dependent rats will self-administer significantly more ethanol during acute withdrawal than rats in a nondependent state. In these studies, Wistar rats are trained with a sweet solution fadeout procedure to self-administer ethanol in a two-lever operant situation in which one lever delivers 0.1 ml of 10% ethanol and the other lever delivers 0.1 ml of water. Nondependent animals typically self-administer doses of ethanol sufficient to produce blood alcohol levels averaging 25–30 mg % at the end of a 30-minute session, but rats made dependent on ethanol with ethanol vapor chambers self-administer three to four times as much ethanol (
Table 1 ). With unlimited access to ethanol during a full 12 hours of withdrawal, the animals will maintain blood alcohol levels above 100 mg %
(39) . When the animals were subjected to repeated withdrawals and ethanol intake was charted over repeated abstinence, operant responding was enhanced by 30%–100% for up to 4–8 weeks postwithdrawal. Similar but even more dramatic results have been obtained with intermittent access to ethanol vapors (14 hours on, 10 hours off)
(41 –
43) . These results suggest an increase in ethanol self-administration in animals with a history of dependence that is not observed in animals maintained on limited access to ethanol of 30 minutes/day. The increase in responding has been hypothesized to be linked to changes in reward set point that invoke the theoretical concepts of tolerance or allostasis.
Stress Hormone Measures in Drug Self-Administration Escalation Models
Previous work has shown a key role for activation of the HPA axis in all aspects of cocaine dependence as measured in animal models
(46) . In an escalation model, when rats were divided into groups according to the doses of cocaine that were available for self-administration, positive correlations were found between presession corticosterone levels and the amount of cocaine self-administered (but only at the lowest level of 0.25 mg/kg and not at 0.50, 1.00, or 2.00 mg/kg)
(34) . Locomotor activity and plasma corticosterone levels before self-administration and food-reinforced lever pressing predicted self-administration along with high response to novelty, but again only with the very lowest dose of cocaine (0.25 mg/kg). There were no correlations between any of these pre-cocaine exposure factors and the subsequent pattern of self-administration in low, moderate, or high doses of cocaine (0.50, 1.00, or 2.00 mg/kg per infusion)
(35) . These findings indicated that predictable individual differences in cocaine self-administration are relevant only when the very low doses are used and are immediately reversed by increasing the doses of cocaine
(34,
35) . Low-dose psychostimulant effects may be related to initial vulnerability to drug use, and activation of the HPA axis may contribute to such vulnerability
(47,
48) . Such low-dose initial actions may parallel the phenomena of locomotor sensitization in which activation of the HPA axis has been shown to faciliate locomotor sensitization
(49) . Also, glucocorticoid antagonists block low-dose cocaine self-administration
(50) and stress-induced reinstatement of cocaine self-administration
(51) .
Prolactin also is a well-recognized stress-responsive hormone involving mechanisms in both hypothalamic and pituitary regions. During each of 5 days of cocaine self-administration (the Kreek group), prolactin levels were significantly lower at the end of the self-administration period than at the beginning, presumably due to the increase in perisynaptic dopamine levels in the midbrain and in the tuberinfundibular dopaminergic system, the site of modulation of prolactin release in mammals. Unlike cortisol levels that persisted as abnormal for several days after cocaine withdrawal, prolactin levels were restored to baseline values within 1 day of withdrawal
(35) . These results suggest the hypothesis that prolactin also may contribute to the dysregulation of neuroendocrine function that characterizes acute withdrawal from psychostimulant drugs.
The Role of CRF in the Motivational Effects of Excessive Drug Taking in Dependent Animals
Long-term exposure to ethanol vapors sufficient to induce dependence produces increases in ethanol self-administration during acute withdrawal and during protracted abstinence
(52 –
54) . Neuropharmacological studies have shown that enhanced ethanol self-administration during acute withdrawal and protracted abstinence can be reduced dose-dependently by intracerebroventricular administration of a competitive CRF antagonist
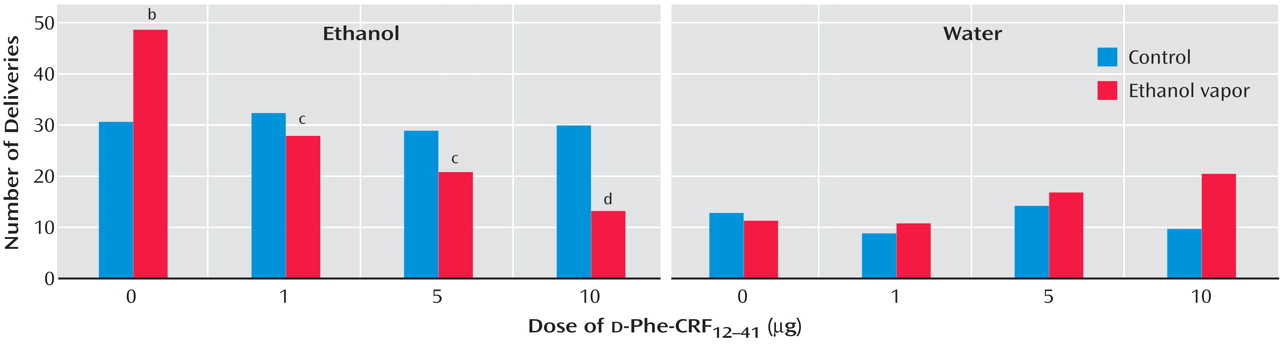
(55) . However, identical doses and administration of CRF antagonists to
nondependent rats had no effect on the self-administration of ethanol. In these studies, male Wistar rats were trained to respond to ethanol (10%) or water in a two-lever free-choice design. The rats received either ethanol vapor (dependent group) or air control (nondependent group). Both groups of rats were tested following a 3–4 week period during which the dependent rats exhibited target blood alcohol levels of 150–200 mg % in alcohol vapor chambers. The rats were tested in 30-minute sessions 2 hours after the dependent rats were removed from the chambers. The results showed that the CRF antagonist
d -Phe-CRF
12–41 dose-dependently decreased operant responding for ethanol in ethanol vapor-exposed rats during early withdrawal but had no effect in air control rats
(55) (
Figure 5 ). The same competitive CRF antagonist also dose-dependently decreased operant responding for ethanol in rats after acute withdrawal (3–5 weeks after vapor exposure) with a history of ethanol vapor exposure but had no effect in air control rats. Similar results have been obtained with direct administration of the competitive CRF
1 /CRF
2 antagonist
d -Phe-CRF
12–41 directly into the amygdala
(42) and with systemic administration of small molecule CRF antagonists
(43,
56) . CRF dysregulation in the amygdala has been observed to persist up to 6 weeks postabstinence
(57) . These results suggest that during the development of ethanol dependence there is a recruitment of CRF activity in the rat of motivational significance that can persist into protracted abstinence. Preliminary results have shown similar effects of systemic administration of CRF
1 receptor antagonists in the escalation in cocaine intake associated with extended access (unpublished study by Specio SE et al.) and in rats with prolonged extended access to heroin (unpublished study by Greenwell TN et al.). The CRF
1 antagonist antalarmin and related CRF antagonists dose-dependently decreased cocaine and heroin self-administration in escalated animals.
Studies on CRF in addiction in humans have been largely limited to CRF challenge studies and measures of CRF in CSF lumbar samples. During short-term and protracted abstinence, human alcoholics showed a blunted cortisol response to CRF
(58,
59) . An elevation of CSF CRF from lumbar samples in human alcoholics during acute withdrawal (day 1) has been observed
(60) . These results are consistent with the animal studies cited above in that they reflect a dysregulation of the HPA axis during protracted abstinence and a potential activation of extrahypothalamic CRF during acute withdrawal.
Hormonal and Brain Changes in Excessive Drug-Taking Associated With Escalation
Animals allowed to self-administer cocaine for 10 hours during their active period showed a blunting of the normal circadian rhythm of cortisol plasma levels by the end of the first day of self-administration of cocaine
(34) . By the third day of cocaine self-administration, there was a complete reversal of the normal cortisol circadian rhythm, with levels of plasma cortisol much higher at the end of the cocaine self-administration session than at the beginning, when they should have been the highest. The blunting of the normal circadian rhythm of cortisol levels continued into withdrawal at 1 and 4 days after the last self-administration of cocaine.
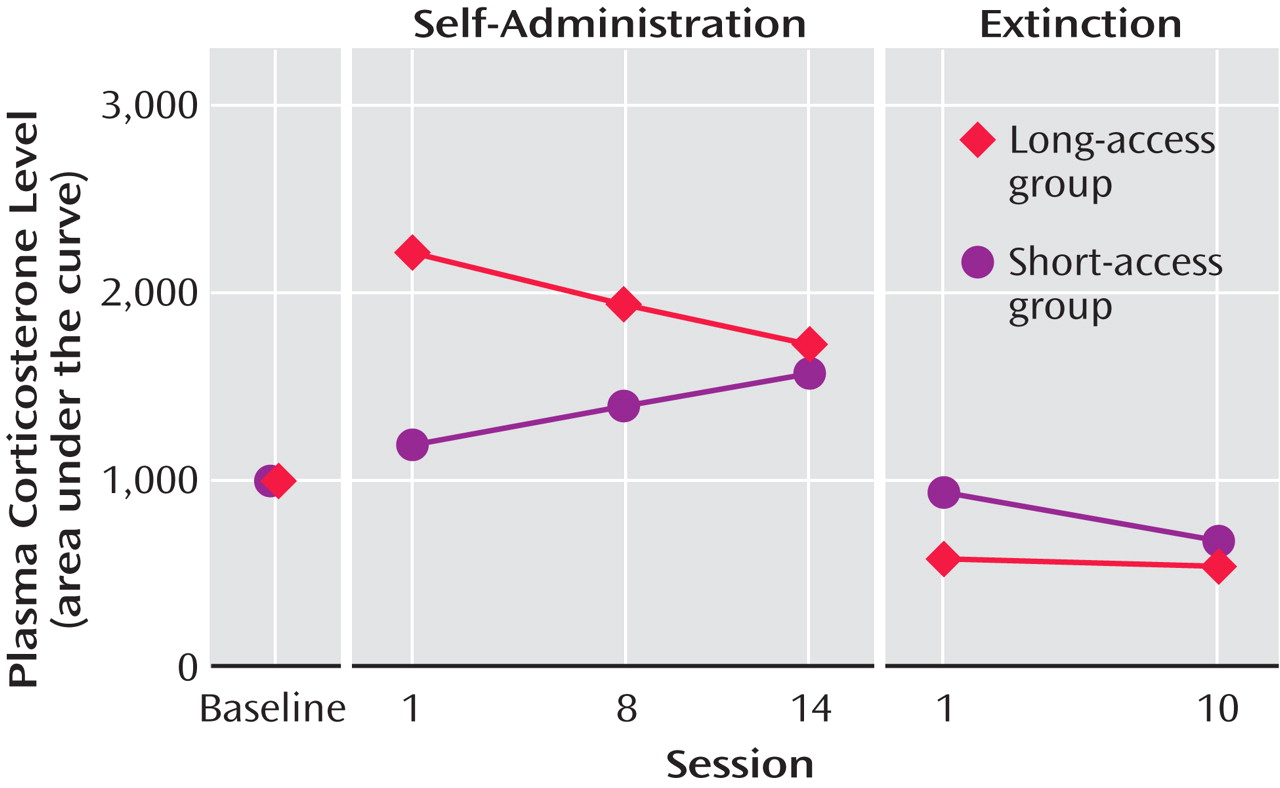
With repeated long-access high-dose cocaine self-administration, daily corticosterone levels, as measured by the area under the plasma corticosterone time curve, progressively decreased (and much further than those that had been observed with 5 days of cocaine self-administration). Similar findings have been made in human long-term cocaine addicts in a clinical laboratory setting
(61) . In contrast, the daily corticosterone area under the plasma concentration time curve in the short-access rats increased across testing, despite a relatively constant rate of self-administration
(36) (
Figure 6 ). In addition, mRNA levels for proopiomelanocortin and the glucocorticoid receptor in the anterior pituitary were significantly lower in the long-access rats than in the short-access rats. However, no differences were found for quantitative measures of CRF mRNA in the amygdala in a direct comparison of short-access long-access low-dose and long-access high-dose animals. Also, corticosterone and hypothalamic CRF mRNA are increased during acute withdrawal from long-term cocaine administration
(62) . Similar decreases in HPA activity have been observed with repeated alcohol administration in a binge model
(63 –
65) . Long-term daily administration of alcohol in a liquid diet showed a decrease in HPA axis activity that persisted up to 3 weeks postabstinence
(66) . Neurochemical studies revealed increases in mRNA for enkephalin in the caudate putamen and increases in the dopamine D
2 receptor in the nucleus accumbens in long-access high-dose rats
(37) . Differences also were not found in preprodynorphin mRNA levels across any of these three groups. In contrast to the findings in the nucleus accumbens, D
2 receptor levels in the anterior pituitary were significantly lower in the long-access rats than in the short-access rats. These findings suggest that at the neuroendocrine and neurochemical levels in the escalation model, there are significant differences in responses of stress-responsive hormones of the HPA axis and some significant differences in quantitatively measured mRNA levels of genes of potential interest both for the reinforcing or rewarding effects and for stress-responsive systems
(37) .
In studies of the escalation of morphine intake, self-regulated dosing of morphine was associated with rapid escalation of total daily consumption but not with alteration in consumption rates. The Kreek group has shown that the m-opioid receptor system is of seminal importance in reward and has a role in modulating the expression of many of the hormonal stress-responsive genes both in the hypothalamus and the anterior pituitary. Studies were conducted to determine the status of the m-opioid receptor activation system focused on two regions related to reward and pain, respectively: the amygdala and the thalamus. Animals in the long-access escalating-dose group showed significantly decreased morphine-stimulated [
35 S]GTPgS binding in membranes prepared from both amygdala and thalamic nuclei compared to the fixed-dose and control groups with cell biological assay studies
(38) . Escalating doses, which mimic the human pattern of morphine or heroin use, are associated with profound alterations in the function of m-opioid receptors. Changes in
N -methyl-
d -aspartic acid (NMDA) NR1 labeling in the tractus solitarius and also changes in AMPA GluR1 subunit labeling on dendrites in the basolateral amygdala were observed in animals subjected to a similar escalation in morphine dosing
(67) . These results suggest that subject-regulated dosing is a useful approach for modeling dose escalation associated with opiate dependence and addiction
(38) .
Allostasis Versus Homeostasis in Dependence and the Role of the Brain Stress Systems
Allostasis is defined as the process of achieving stability through change. An
allostatic state is a state of chronic deviation of the regulatory system from its normal (homeostatic) operating level. Allostasis originally was formulated as a hypothesis to explain the physiological basis for changes in patterns of human morbidity and mortality associated with modern life
(68) . High blood pressure and other pathology was linked to social disruption by brain-body interactions.
Allostatic load is the cost to the brain and body of the deviation accumulating over time and reflecting in many cases pathological states and accumulation of damage. Using the arousal/stress continuum as their physiological framework, Sterling and Eyer
(68) argued that homeostasis was not adequate to explain such brain-body interactions. The concept of allostasis has several unique characteristics that lend it more explanatory power. These characteristics include a continuous reevaluation of the organism’s need and continuous readjustments to new set points depending on demand. Allostasis can anticipate altered need, and the system can make adjustments in advance. Allostasis systems also were hypothesized to use past experience to anticipate demand
(68) .
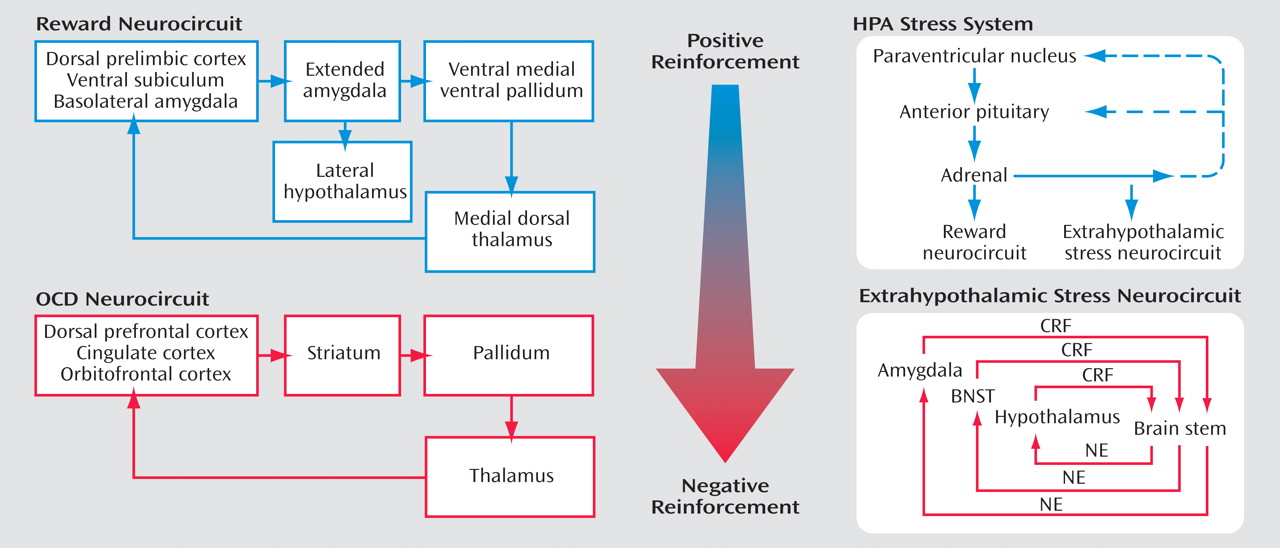
Self-administration of drugs may initiate a cascade of stress responses that play a role in allostatic, as opposed to homeostatic, responses
(69) (
Figure 7 ). An acute binge of drug-taking beyond that of limited access produces an activation of the HPA axis, which activates or prolongs the activation of the brain reward systems and which, during a more prolonged binge, activates the brain stress systems. However, the neuroendocrine aspect of the stress response also has its capacity blunted, either by negative feedback or by depletion or both. Acute withdrawal from drugs of abuse produces opponent process-like changes in reward neurotransmitters in specific elements of reward circuitry associated with the ventral forebrain, as well as recruitment of brain stress systems that motivationally oppose the hedonic effects of drugs of abuse.
From the drug addiction perspective, allostasis is the process of maintaining apparent reward function stability through changes in reward and stress system neurocircuitry. The changes in brain and hormonal systems associated with the development of motivational aspects of withdrawal are hypothesized to be a major source of potential allostatic changes that drive and maintain addiction. The neuropharmacological contribution to the altered set point is hypothesized to involve not only decreases in reward function, including dopamine, serotonin, and opioid peptides, but also recruitment of brain stress systems such as CRF. All of these changes are hypothesized to be focused on a dysregulation of function within the neurocircuitry of the basal forebrain associated with the extended amygdala (central nucleus of the amygdala and bed nucleus of the stria terminalis). The present formulation is an extension of the opponent process of Solomon and Corbit
(70) to an allostatic framework with a hypothesized neurobiologic mechanism.
The initial experience of a drug with no prior drug history shows a positive hedonic response ( a-process ) and a subsequently minor negative hedonic response ( b-process ), each represented by increased and decreased functional activity of reward transmitters, respectively. The b-process also is hypothesized to involve recruitment of brain stress neurotransmitter function. However, insufficient time between readministering the drug to retain the a-process and limit the b-process leads to the transition to an allostatic reward state, as has been observed in the escalation of cocaine, methamphetamine, heroin, and ethanol intake in animal models. Under conditions of an allostatic reward state, the b-process never returns to the original homeostatic level before drug-taking resumes. This dysregulation is driven in part by an overactive HPA axis and subsequently overactive CNS CRF system and thus creates a greater and greater allostatic state in the brain reward systems and, by extrapolation, a transition to addiction.
Thus, in escalation situations (extended access), the counteradaptive opponent process does not simply balance the activational process ( a-process ) but, in fact, shows residual hysteresis. The results with cocaine escalation and brain reward thresholds provide empirical evidence for this hypothesis. The neurochemical, hormonal, and neurocircuitry changes observed during acute withdrawal are hypothesized to persist in some form even during postdetoxification, defining a state of “protracted abstinence.” The results described above are shedding some light on a potential role for the CRF brain and hormonal stress systems in protracted abstinence.
An allostatic view of drug addiction thus provides a heuristic model with which to explore residual changes following drug binges that contribute to vulnerability to relapse
(1,
15,
71) . What should be emphasized is that from an allostatic perspective, the allostatic load in addiction is a persistent state of stress in which the CNS and HPA axis are chronically dysregulated. Such a state provides a change in baseline such that environmental events that would normally elicit drug-seeking behavior have even more impact. Recently, direct evidence for this has been documented in human cocaine addicts
(72,
73) . Much work remains to define the neurochemistry and neurocircuitry of this residual stress state, and such information will be the key to its reversal and will provide important information for the prevention and treatment of drug addiction. The hypothesis suggested here is that the neurobiological bases for this complex syndrome of protracted abstinence may involve subtle molecular and cellular changes in stress system neurocircuitry associated with the extended amygdala.
The present review has focused on the CRF system and the HPA axis because this system is known to be a critical modulator of both hormonal and behavioral responses to stressors
(74 –
79) . However, there are many more stress regulatory systems in the brain that may also contribute to the allostatic changes hypothesized to be critical to the development and maintenance of motivational homeostasis, including norepinephrine
(17), neuropeptide Y
(80,
81), nociceptin
(82), orexin, and vasopressin
(83) . These same neurochemical systems may be involved in mediating anxiety disorders and other stress disorders, and the elucidation of the role of the stress axis in drug dependence may also provide insights into the role of these systems in other psychopathology.