Discussion
Although medical causes must always be ruled out in the case of new-onset mania, this is particularly important in the elderly. Older patients with new-onset mania are more than twice as likely to suffer from a comorbid neurological disorder
(3), including silent cerebral infarcts (65% in new-onset versus 25% in chronic illness)
(4) . In addition to a standard history and a physical, these patients require a thorough neurologic examination, neuroimaging, and other selected tests
(5) . Reported neurological causes of late-onset mania include stroke, tumors, epilepsy, Huntington’s disease and other movement disorders, multiple sclerosis and other white matter diseases, head trauma, infections such as neurosyphilis, Creutzfeldt-Jakob disease, and frontotemporal dementia (for a review, see references 6–8).
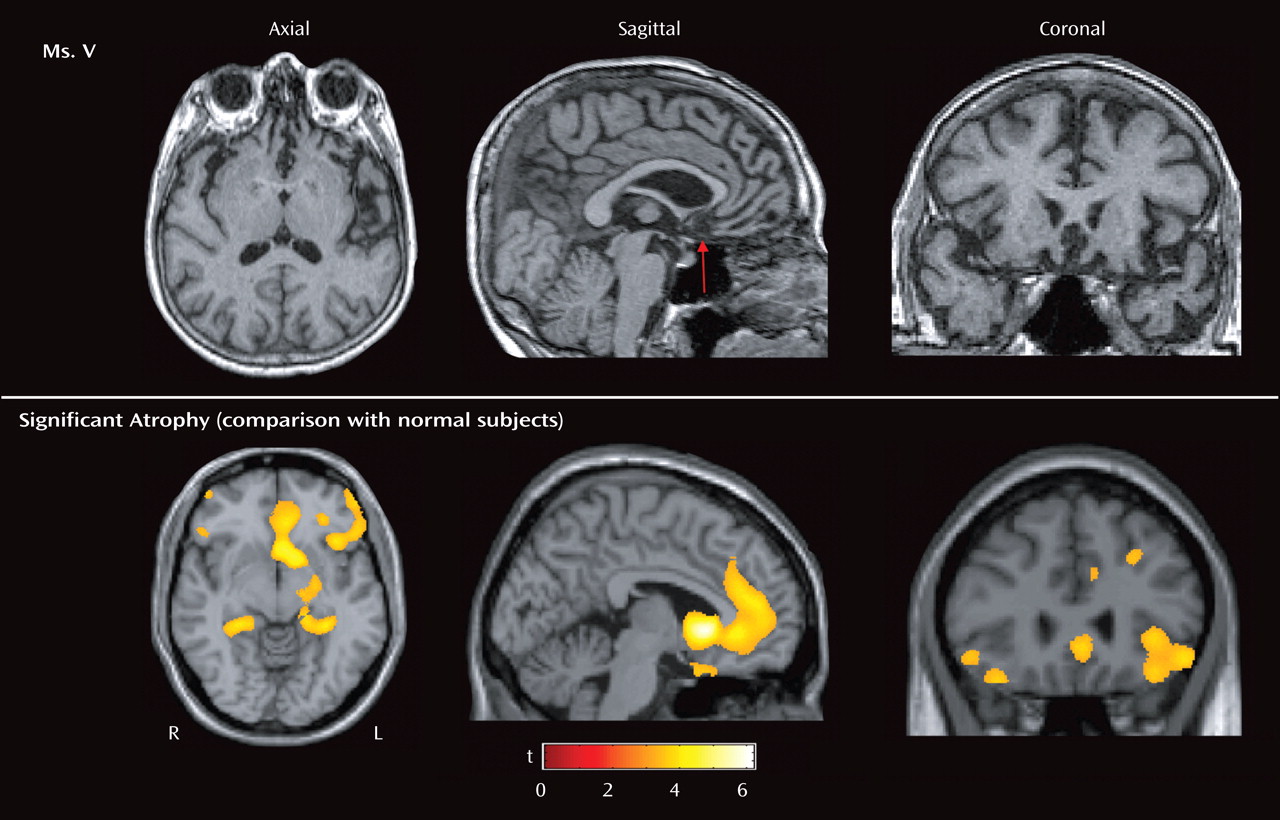
Ms. V’s symptoms of irritability, pressured speech, expansiveness, decreased need for sleep, impulsivity, and psychomotor agitation all suggested mania. Clues that she had a neurodegenerative disease included her age as well as the chronic and progressive nature of her symptoms. The profound loss of sympathy and empathy for others, along with severe disinhibition and the repetitive motor behaviors (e.g., checking locks), suggested frontotemporal dementia. Frontotemporal dementia is a progressive neurodegenerative disease that affects the frontal and temporal cortices, has several clinical subtypes (for a comprehensive review, see references
9 and
10 ), and is a common cause of dementia in patients younger than 65
(11) . The frontal lobe variant of frontotemporal dementia (also known as behavioral variant or social/executive disorder variant) is characterized by insidious behavioral and personality changes, and often the initial presentation lacks any clear neurological signs or symptoms. Key features include apathy, disinhibition, loss of sympathy and empathy for others, repetitive motor behaviors, overeating, and frontal/executive deficits on neuropsychological testing. Because of these features, in one series
(12), patients with frontotemporal dementia were more likely to initially present to a psychiatrist than to a neurologist and often received erroneous psychiatric diagnoses before frontotemporal dementia was correctly diagnosed.
Affective blunting, loss of insight and social awareness, lack of concern about the disease process (so-called
la belle indifference ), coldness, and lack of empathy for family and friends were prominent symptoms in Ms. V and are common symptoms in frontal lobe variant frontotemporal dementia
(9,
13,
14) . Furthermore, damage to the ventromedial portion of the frontal cortex, as seen in Ms. V, leads to disinhibition, poor impulse control, and antisocial behavior in patients with discrete lesions
(15) and frontotemporal dementia
(16,
17) . Similarly, atrophy to a frontal-temporal circuit, including the ventromedial frontal cortex in frontotemporal dementia, has been linked to loss of empathy
(18) . Patients with frontotemporal dementia with asymmetric atrophy of the nondominant frontal lobe have also been shown to exhibit dramatic alterations in their self as defined by changes in political, social, or religious values
(19) . Stereotyped behaviors, such as compulsive cleaning, pacing, and collecting are also common in frontotemporal dementia; in addition to often presenting as acute mania (e.g., reference
20 ), patients with frontotemporal dementia frequently present solely with compulsive symptoms
(21) . In dementia, these repetitive motor behaviors are associated with atrophy in the right supplementary motor region of the frontal lobes
(17) . Ms. V was also apathetic (e.g., not seeking a new job), which is a common symptom in frontotemporal dementia associated with atrophy of the anterior cingulate and medial frontal lobes
(22) . This can be confused with depression, which is not common in frontotemporal dementia; patients with frontotemporal dementia are apathetic and emotionally withdrawn but not sad or suicidal. Often (up to one-third) of patients with frontotemporal dementia exhibit euphoria, which can take the form of elevated mood, inappropriate jocularity, and exaggerated self-esteem that can be indistinguishable from hypomania or mania (for a review, see reference
23 ). Gluttonous overeating is also common in frontotemporal dementia
(24 –
26) and appears to be due to disruption of a right-side orbitofrontal-insular-striatal circuit
(27,
28) . Similarly, an exaggerated craving for carbohydrates has also been demonstrated in frontotemporal dementia by questionnaire
(24,
25,
29) and laboratory
(27) studies. The absence of these symptoms in Ms. V may be due to relative sparing of her right ventral insula and striatum.
Therapeutically, serotonin reuptake inhibitors are common first-line agents given the well-described serotonergic deficits in frontotemporal dementia
(29,
30) . A recent meta-analysis demonstrated these agents are effective in the treatment of frontotemporal dementia
(31) (but see reference
32 ). Atypical antipsychotics and mood stabilizers can also be used to control the behavioral symptoms of frontotemporal dementia
(33), although data on their efficacy is limited. Furthermore, patients with frontotemporal dementia are known to have dopaminergic deficits
(31) and are particularly prone to developing extrapyramidal side effects
(34) . Although acetylcholine deficits are well described in Alzheimer’s disease
(35) and have led to successful therapeutic use of acetylcholinesterase inhibitors in this disease
(36,
37), the acetylcholine system in frontotemporal dementia appears to be relatively intact
(30,
31) . In our and others’ clinical experience, acetylcholinesterase inhibitors are ineffective in frontotemporal dementia and contraindicated since they can worsen patients’ agitation, anxiety, behavioral alteration, and restlessness
(34,
38, but see reference
39 ). The
N -methyl-
d -aspartic acid (NMDA) antagonist memantine has also been tried, and one small, open-label, uncontrolled study has shown it may be helpful for behavioral disturbances in frontotemporal dementia
(40) .
Historically, the clinical features of what would first be termed Pick’s disease and later called frontotemporal dementia were first delineated by the German psychiatrist Arnold Pick in 1892
(41) . He described a 71-year-old gentleman with progressive aphasia, apraxia, and behavior change in association with frontotemporal atrophy. In 1926, pathologists found that round intraneuronal inclusions, when, stained with silver, were characteristic of this illness
(42) . In honor of Arnold Pick, these inclusions were later called Pick bodies
(43) . More recently, these inclusions were found to contain aggregations of insoluble hyperphosphorylated tau protein. Thus, frontotemporal dementia came to be grouped in a class of neurodegenerative diseases called the tauopathies (others include semantic dementia, primary progressive aphasia, progressive supranuclear palsy, and corticobasal degeneration).
The understanding of frontotemporal dementia has improved over the past 15 years. During this time, large autopsy studies have found that the “classic” tau pathology of frontotemporal dementia is actually absent in 60% of patients with clinically diagnosed frontotemporal dementia. Instead, many of these patients’ brains display ubiquitin-immunoreactive neuronal cytoplasmic inclusions and neuritic changes in the cerebral cortex and the hippocampus
(44,
45) . This neuropathologic pattern was labeled frontotemporal lobar degeneration with ubiquitin inclusions
(46) . Until very recently, both the identity of the ubiquinated protein that comprises the inclusions and the underlying pathophysiology of frontotemporal lobar degeneration with ubiquitin inclusions have remained a mystery.
A clue to better understanding frontotemporal lobar degeneration with ubiquitin inclusions lay in the fact that overall, frontotemporal dementia has a strong genetic component with up to 20% of cases showing a highly penetrant, autosomal dominant pattern of disease transmission
(47 –
49) . In 1998, in a subset of patients with frontotemporal dementia demonstrating classic tau pathology, mutations in the microtubule-associated protein tau gene on chromosome 17q21 were identified
(50) . These mutations remain relatively uncommon, even in familial forms of frontotemporal dementia
(51) .
In 2006, two back-to-back articles in
Nature reported that mutations in the gene for a widely expressed growth factor called progranulin caused multiple cases of familial and even some cases of sporadic frontotemporal lobar degeneration with ubiquitin inclusions
(52,
53) . The investigators described a variety of frameshift mutations, initiation codon, and nonsense mutations, all leading to a null mutation in one of the two progranulin alleles, resulting in haploinsufficiency (i.e., loss of functional progranulin). In an apparent coincidence, the progranulin gene is also found in chromosome region 17q21, only 1.7 Mb upstream of microtubule-associated protein tau gene, explaining why a number of frontotemporal dementia families who had been shown to have mutations linked to the chromosome 17q21 region lacked any abnormalities in their microtubule-associated protein tau gene
(54) . Overall, progranulin mutations account for approximately 10% of frontotemporal dementia cases and approximately 20% of cases with a family history
(53,
55,
56) .
Progranulin is known to play systemic roles in development, wound repair, and inflammation through its effects on cell cycle progression and motility
(57 –
59) . In the CNS, it is most highly expressed in pyramidal neurons and activated microglia, but its specific role in the brain remains unknown
(60) . Of interest, when progranulin is overexpressed, it has been linked to tumor genesis, possibly through induction of vascular endothelial growth factor
(58,
59,
61), whereas in circumstances of haploinsufficiency, it leads to neurodegeneration.
Frontotemporal dementia patients are approximately evenly divided between those with tau or frontotemporal lobar degeneration with ubiquitin inclusions. In those with the frontotemporal lobar degeneration with ubiquitin inclusions histopathologic pattern, one subset has lentiform intranuclear neuronal inclusions. It is this subset of frontotemporal lobar degeneration with ubiquitin inclusions patients with both neuronal cytoplasmic inclusions and lentiform intranuclear neuronal inclusions that have been found to have mutations in the progranulin gene. Of importance, neuronal cytoplasmic inclusions and lentiform intranuclear neuronal inclusions do not stain for progranulin and instead were recently found to stain for another protein, TAR DNA binding protein-43
(62) . This is consistent with the finding that the progranulin mutations that cause frontotemporal dementia are null alleles leading to a dearth of wild-type progranulin and little to no production of mutant progranulin protein or sometimes even its mRNA. In addition, the discovery that the nuclear protein TAR DNA binding protein-43 is found in the inclusions of all frontotemporal lobar degeneration with ubiquitin inclusions subtypes (i.e., in both neuronal cytoplasmic inclusions and lentiform intranuclear neuronal inclusions) suggests that a common pathogenic pathway unites frontotemporal dementia’s disparate histopathologic forms.
There is great clinical variability, even within families sharing the same progranulin mutation
(63,
64) . Also, a recent study comparing patients with frontotemporal dementia with progranulin mutations to those with microtubule-associated protein tau gene mutations found few consistent phenotypic differences between the two groups
(65) . Previous work has shown that 15% of patients with frontotemporal dementia have clinical and electromyograph findings consistent with amyotrophic lateral sclerosis
(66) . However, in a large genetic survey, no patients with frontotemporal dementia-amyotrophic lateral sclerosis were found to have progranulin mutations, despite the fact that frontotemporal dementia-amytrophic lateral sclerosis patients also develop a frontotemporal lobar degeneration with ubiquitin inclusions histopathology
(67) . Rather, the motor difficulties experienced by patients with frontotemporal dementia with progranulin mutations more frequently take the form of mild parkinsonism
(65) .
Can this clinical variability even among patients with frontotemporal dementia with progranulin mutations be explained by the many different progranulin mutations already identified? This explanation appears unlikely in light of the considerable intrafamily phenotypic variability as well as the fact that although many progranulin mutations have been described, they all seem to result in the same endpoint, which is decreased levels of wild-type progranulin. Rather, the phenotypic variability most likely arises out of the variable anatomic distribution of the frontotemporal dementia pathology, which likely results from unknown genetic or environmental modifiers.
The recent advances in our understanding of the genetics and pathophysiology of frontotemporal dementia stimulate great hope for future research and therapies. In particular, the finding that insufficient levels of progranulin can result in frontotemporal dementia suggests a potential simple replacement treatment modality that awaits further investigation.