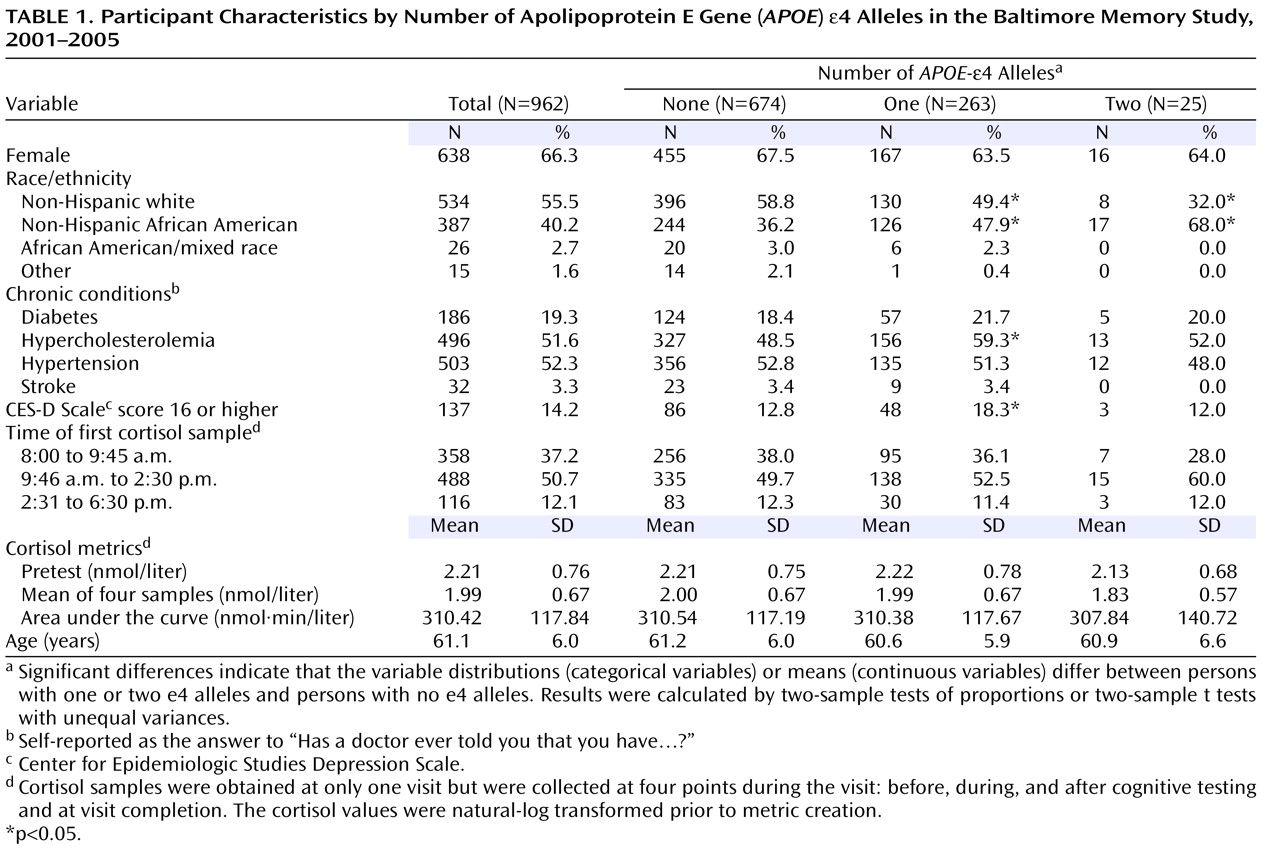
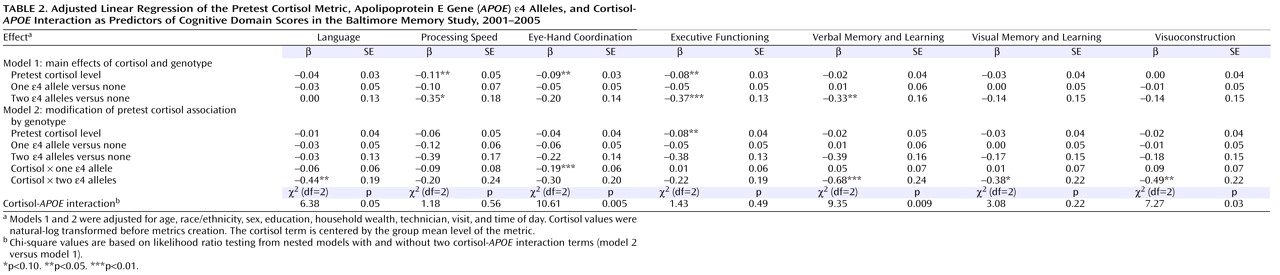
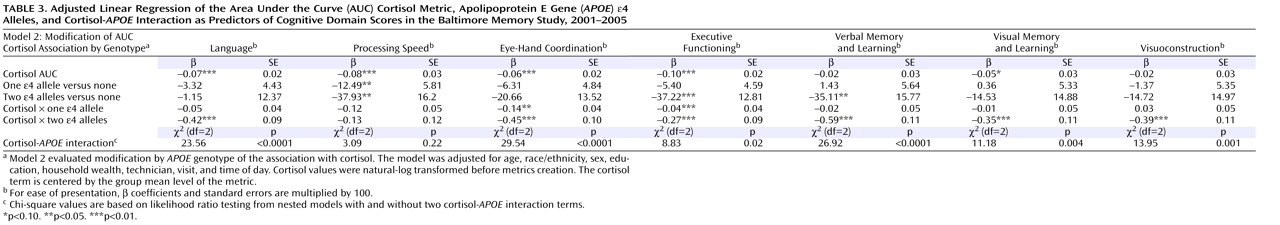
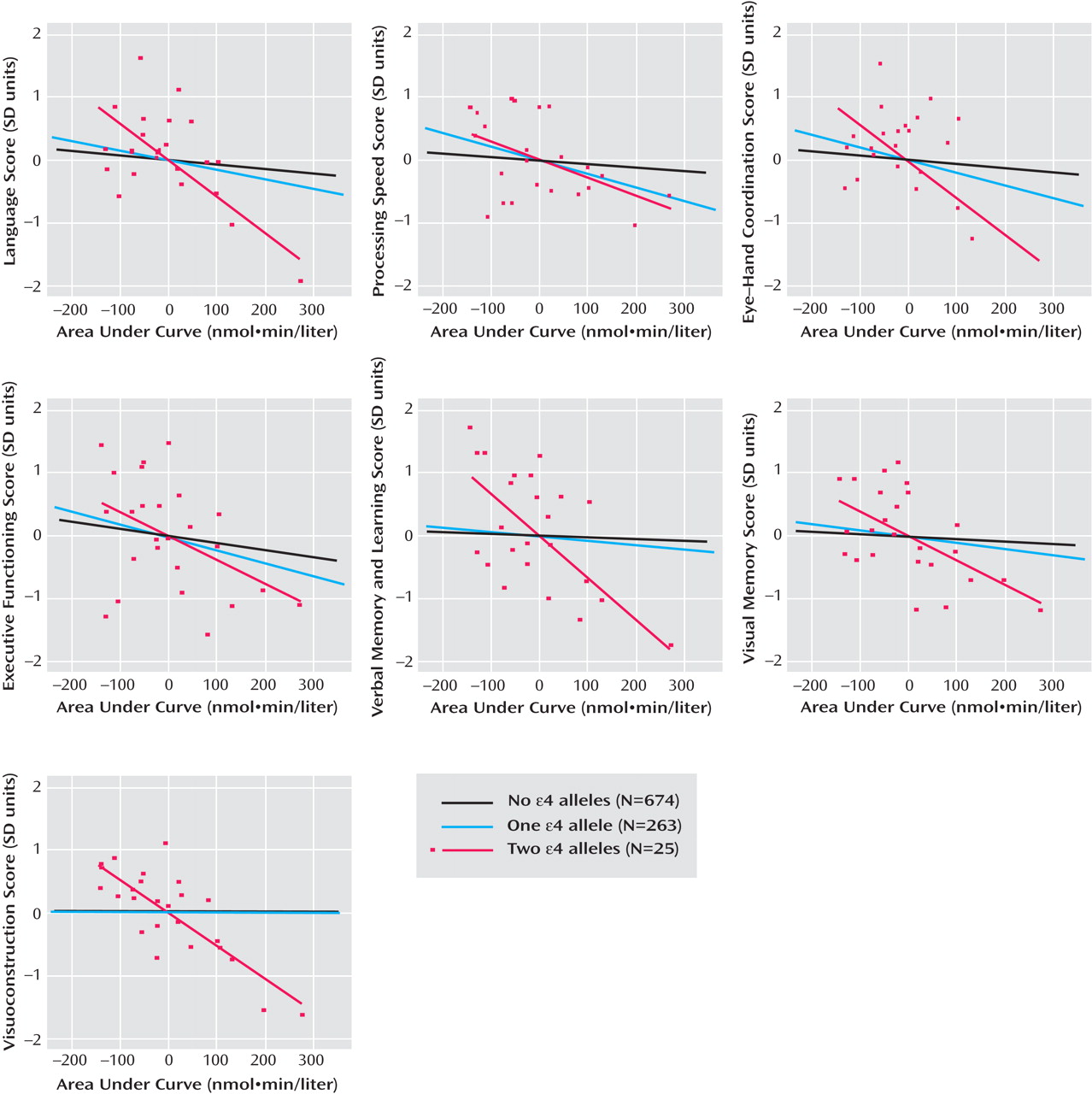
Chronic psychosocial stress has been implicated as a factor contributing to cognitive aging. Animal studies suggest that repeated exposure to psychosocial hazards such as restraint stress or subordinate hierarchical status can cause neuroanatomical changes
(1), including inhibition of adult neurogenesis in the dentate gyrus
(2) and dendritic atrophy in the hippocampus
(3) and medial prefrontal cortex
(4) . This damage is partially attributable to HPA axis dysregulation characterized by excesses in glucocorticoid production and changes in the diurnal pattern of secretion
(5,
6) . Observational studies in older humans indicate that elevation in cortisol (the principal human glucocorticoid) is associated with smaller hippocampal volume
(7) and is a risk factor for greater decline in global cognition, verbal memory, and executive functioning
(8 –
10) .
It is reasonable to expect that genetic factors may influence individual vulnerability to the adverse effects of stress, and one candidate of immediate interest is the gene for apolipoprotein E (
APOE ). Three alleles of
APOE (e2, e3, ε4) code for three protein isoforms that have substantial involvement in lipid metabolism and neurobiology
(11) . The ε4 allele increases risk for late-onset familial and sporadic Alzheimer’s disease in a dose-dependent manner
(12,
13) . The ε4 genotype may influence some domains of cognitive performance in nondemented adults, although effects appear small. A meta-analysis of 38 studies found modest associations of
APOE -ε4 with decrements in global cognition, episodic memory, and executive functioning
(14) . A large study showed greater longitudinal decline for ε4 carriers in verbal memory and processing speed; however, the differences in decline by genotype were relatively small
(15) . Aged healthy ε4 carriers have greater risk of hippocampal atrophy
(16) as well as reduced white matter integrity in the hippocampus, medial temporal lobe, and corpus callosum
(17) . It is noteworthy that epidemiologic evidence suggests that the ε4 allele also amplifies the effects of multiple risk factors for cognitive dysfunction, including head injury, lead exposure, diabetes, peripheral vascular disease, atherosclerosis, hypercholesterolemia, elevated homocysteine, and low vitamin B
12 (15,
18 –
22) .
Discussion
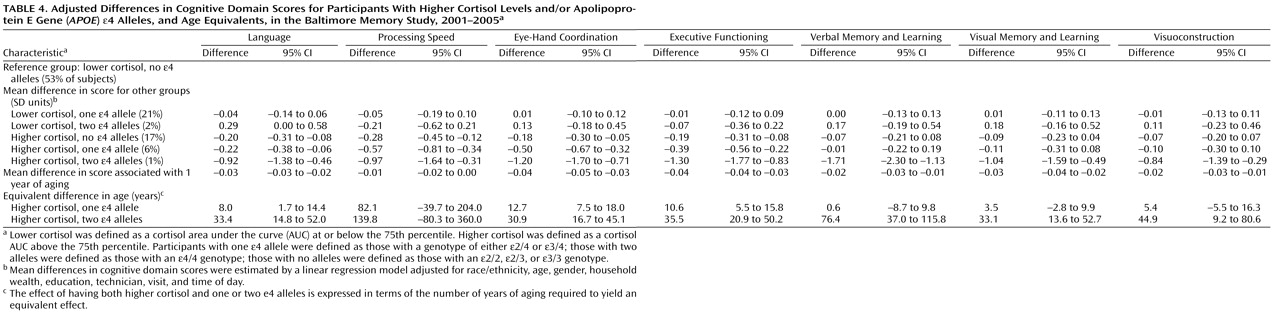
Previously, we found that higher levels of the pretest, mean, and AUC cortisol metrics were associated with worse cognitive performance in a population of community-dwelling older adults. The present findings from the same study population suggest that APOE -ε4 genotype modifies the relation between cortisol and cognitive function such that the slopes of the adverse relations are steeper in the presence of the ε4 allele.
We observed cortisol- APOE interactions in a broad range of domains, including language, eye-hand coordination, executive functioning, verbal memory and learning, visual memory, and visuoconstruction. While memory-related associations with cortisol have been well documented, there are few prior studies of nonmemory associations. Therefore, we cannot speculate about the significance of domain-specific associations. However, since glucocorticoid receptors are well distributed throughout the brain and APOE genotype influences multiple aspects of brain physiology, we believe that the observed interaction in multiple cognitive domains is biologically plausible. The strongest interactions were observed with the AUC metric, with statistically significant interactions observed in six of seven domains. This may be because AUC, in contrast to pretest and mean values, reflects both nonchallenge and challenge cortisol levels as a more comprehensive indicator of HPA axis activity.
It was interesting that there were more significant associations, and the magnitudes of the associations were larger, for the cortisol-
APOE -ε4 interactions than for ε4 alone. There were several associations of
APOE genotype with cognitive decrements, especially among persons with two ε4 alleles, and these findings were generally consistent with prior evidence
(14,
15) . However, the data suggest that among our study subjects the independent influence of
APOE -ε4 genotype on cognitive function may be less important than the combined roles of the genotype and environmental exposures such as stress (as measured by the cortisol metrics), an example of gene-environment interaction.
Our findings are supported by a recent study showing that persons with high self-reported stress levels and any
APOE -ε4 alleles performed worse on several memory tasks
(28) . Because the relation of self-reported chronic stress to the underlying biology of the HPA axis is somewhat unclear, the assessment of HPA axis functioning in the present study provides additional insight into possible biological mechanisms, discussed in the following. We also extend these findings through use of a broader cognitive battery and a larger and younger population-based sample, in which cognitive decline may not be as readily evident.
Population stratification is a possibility, given the strong representation of African Americans in our study group and the higher proportion of African American individuals with an ε4 allele. We addressed this concern by conducting stratified analyses separately for whites and African Americans. We analyzed a model with an indicator for the ε4 allele and its cross-product with cortisol along with model 1 covariates, and we compared the results for the two race/ethnicity groups. While the patterns of statistical significance changed because of the decreased sample size in each stratum, the magnitudes and signs of coefficients for cross-product terms were qualitatively similar by race/ethnicity. This suggests that population stratification is unlikely to account for our findings.
Our results have several limitations. First, the subjects were not clinically assessed for dementia. Estimates place the prevalence of dementia at age 60–64 at less than 1% and at age 65–69 at less than 2%
(29) . In our relatively young community-dwelling and population-based sample, it is unlikely that the prevalence of dementia would be high enough to influence our results. In addition, subjects were not excluded from the study on the basis of drug or medication use that could have altered cognition. However, our previous analysis showed that adjustment for alcohol, tobacco, recreational drug use, and medication use (antidepressants, anxiety medications, and hormone replacement therapy) did not substantially alter the main effect associations of the cortisol metrics with cognitive performance
(23) . Another limitation is the collection of cortisol across an approximately 10-hour time range. However, prior studies suggest that comparable HPA axis responses to psychosocial challenges can be reliably measured in the morning and afternoon
(30) . Furthermore, our analysis of cortisol main effects showed no indication that the results varied between participants sampled in the morning and afternoon
(23), and the time of sampling did not vary by genotype. We therefore do not believe that sampling at different times of the day accounts for our results.
Because our analysis was cross-sectional, we cannot be certain about the temporal relations of our measurements of cortisol and cognitive function. However, several lines of evidence allow us to infer that the HPA axis dysregulation we believe the cortisol metrics measure can be placed before the measured cognitive function. Although cortisol was assessed at the same time as cognitive testing, we previously found that 1) adjustment for perceived distress at the time of sampling did not alter the associations of cortisol and cognitive performance and 2) the rate of change in cortisol from the pretest to the second measurement shortly after the peak difficulty in cognitive testing (slope from cortisol samples 1-2) was not associated with cognitive performance
(23) . These observations indicate that associations between cortisol and cognitive function likely were not due to acute, short-latency effects but rather longer-term effects of cortisol that preceded the study visit. This is in accordance with longitudinal studies
(8 –
10) that provide evidence for the chronic effects of excess glucocorticoids on cognitive decline. Because temporality in the case of a genetic polymorphism is not a concern, we believe that elevated cortisol and
APOE genotype-specific effects both temporally precede any decrements in cognitive function in older age.
While the roles of both
APOE and cortisol in the pathogenesis of cognitive dysfunction are still not well understood, several plausible biological mechanisms can be proposed for the cortisol-
APOE interaction.
APOE genotype may modify the physiological consequences of HPA axis activity. For instance, the effects of environmental stressors
(31) and administered corticosterone
(32) on cognitive performance are conditional on
APOE genotype in mice. In addition,
APOE genotype may lower the neuronal threshold required for cortisol or neurotoxicants to produce neurodegenerative changes. Experimental stress conditions such as restraint or corticosterone injection produce dendritic atrophy of the hippocampus in rats, which is reversible with either termination of stress-inducing conditions or pharmacological treatment
(33,
34) . Apolipoprotein E has isoform-specific effects on neurite remodeling, and its significant involvement with proliferation, repair, and remyelination can affect recovery from neurotoxic insults
(11) . Thus, the ability to recover from potentially reversible detrimental effects of cortisol, especially in the aging brain, may differ by
APOE genotype.
It is also possible that cortisol may increase susceptibility of the brain to adverse events, such as the structural and functional changes mediated by apolipoprotein E. Corticosterone has been shown to exacerbate damage to hippocampal cell cultures by glutamate, FeSO
4, and amyloid beta toxicities
(35) . In addition, elevated cortisol reduces hippocampal glucose metabolism in elderly persons
(36) . Given that older, nondemented ε4 carriers exhibit cerebral glucose hypometabolism in the parietal, temporal, and posterior cingulate cortices
(37), one possible mechanism for the interaction of
APOE and cortisol takes place through coincident energetics disruption and the resulting impairment of neuronal calcium regulation
(38) .
Until this study’s findings are replicated, discussion of relevant clinical implications may be premature. Nevertheless, it is intriguing that both glucocorticoids and
APOE have been implicated as playing central roles in the disease process of Alzheimer’s disease. Experiments in a mouse model have shown that stress-level glucocorticoid administration increases production of amyloid beta and accumulation of tau pathology
(39), suggesting that the higher cortisol levels often found in patients with Alzheimer’s disease are not merely a consequence of the disease. Additionally, patients’ higher plasma cortisol levels are associated with accelerated progression of dementia symptoms and more rapidly decreasing cognitive performance
(40) . It is possible that the HPA axis and
APOE genotype may have joint effects in the disease process of Alzheimer’s disease.
In summary, the results from this study indicate that APOE -ε4 genotype may influence vulnerability to the effects of HPA axis dysregulation on cognitive function. Given that the cortisol metrics may capture HPA axis dysregulation and that such dysregulation could result from chronic stress, it is plausible that the results here are evidence of a gene-environment interaction concerning the adverse effects of the environment and its psychosocial hazards for the cognitive function of older persons.