Huntington's disease is a progressive, dominantly inherited neurodegenerative disorder characterized by motor and cognitive symptoms. It is caused by a mutation in one copy of the Huntingtin (
HTT) gene, comprising an expanded version of a polymorphic CAG trinucleotide repeat (
1,
2). Normal chromosomes possess CAG alleles ranging from six to 34 units. Repeat expansions of 40 units or more are fully penetrant and lead to onset of overt clinical symptoms, with age at onset inversely correlated with the repeat length, whereas repeats of 36–39 units may exhibit reduced penetrance (
3). Recent reports underscore the prevalence of psychiatric symptoms in Huntington's disease as well, particularly depression, irritability, and impaired impulse control, which may be present early in the disease course (
4) or even precede the characteristic chorea by 10 years or more (
5). In one study of presymptomatic Huntington's disease repeat carriers, for example, study subjects displayed greater levels of irritability as well as cognitive symptoms (
6).
Studies of the pathophysiology of major depressive disorder have revealed associated changes in striatal perfusion at rest (
7) and during reward processing (
8–
10) and learning tasks (
11). Recently, direct stimulation of striatum has shown evidence of efficacy in difficult-to-treat major depression (
12). More broadly, high rates of depression are observed across basal ganglia diseases (
13,
14), including caudate stroke (
15). Likewise, a growing body of evidence suggests glutamatergic abnormalities in major depression (
16), and glutamatergic drugs may exhibit antidepressant effects (
17). Thus, both in clinical presentation and in pathophysiology, similarities between Huntington's disease and major depressive disorder are apparent.
Recent studies suggest that a small but definable proportion of apparent cases of psychiatric disorders such as schizophrenia may be accounted for by rare copy-number variations. One example is the 22q11 deletion associated with velocardiofacial syndrome, present in multiple studies of clinical samples of schizophrenia patients (
18,
19). While it is known that even "presymptomatic" carriers of the Huntington's disease risk allele may exhibit mood symptoms, it has been assumed that carriers would still be extremely rare in clinical populations with major depression. To our knowledge, only one small study has examined the
HTT CAG repeat in patients diagnosed with major depression, and it found no expanded alleles among 16 patients (
20). Thus, from a clinical perspective, it is unknown how commonly individuals presenting with major depression carry risk alleles for Huntington's disease.
To better characterize the prevalence of HTT CAG repeats in clinical populations with major depression, we first examined HTT CAG repeat length in two cohorts of affected individuals, including one drawn from the Sequential Treatment Alternatives to Relieve Depression (STAR*D) cohort, and in comparison subjects. We hypothesized that expanded HTT alleles would be overrepresented among individuals with major depression relative to comparison subjects. To attempt to replicate initial findings, we then examined repeat length in a third cohort of patients with major depression and comparison subjects.
Method
Clinical Cohort 1
As part of a study of genetic influences on neuroimaging phenotypes, the Phenotype Genotype Project (
21), three groups of individuals were recruited through newspaper advertisements or referrals from psychiatric clinics: individuals with major depressive disorder, individuals with cocaine use disorders, and a healthy comparison group matched for age, sex, education, and ethnicity. All participants underwent a clinical interview that included the Structured Clinical Interview for DSM-IV Axis I Disorders (SCID;
22). Race was determined by self-identification on a standardized form (
23). Participants also completed the Inventory of Depressive Symptomatology–Self Report (
24,
25) and the Edinburgh Handedness Inventory (
26). Participants had to be 18–55 years of age and have no current or lifetime DSM-IV axis I disorder or major medical illness known to influence brain structure or function, including neurologic disease, HIV, and hepatitis C. All participants provided written informed consent, and the protocol was approved by the institutional review board of Massachusetts General Hospital. In the major depression group (N=34), the mean age was 38.4 years (SD=11.2), all were Caucasian, and 47.1% were women. In the comparison group (N=74), the mean age was 34.8 years (SD=11.9), all were Caucasian, and 46.0% were women.
Clinical Cohort 2
The Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study was a multicenter study conducted in the United States with more than 4,000 individuals who presented to primary or specialty care clinics for treatment of depression. Details of the STAR*D protocol and clinical results have been presented previously (
27,
28). DNA was collected for 1,932 of the participants and was made available to us in an anonymized form by the National Institute of Mental Health; DNA from 1,493 self-reported Caucasian participants was used in the present analysis. In this group, 61.0% were women, and the mean age was 42.7 years (SD=13.5). The STAR*D clinical and genetics protocols were approved by the institutional review boards of participating institutions. Because the DNA was provided in anonymized form, the Massachusetts General Hospital institutional review board determined that the study protocol did not represent human subjects research.
Determining an appropriate comparison group for this study was complicated by the late onset of Huntington's disease and by the fact that, to our knowledge, the HTT CAG repeat has not been examined in large non-Huntington's cohorts. Therefore, because Huntington's disease is a relatively uncommon dominantly inherited disorder (1 in 10,000), we used as an initial comparator for this cohort CAG repeat frequency on the second chromosome (i.e., the one with the shorter repeat) in large cohorts of patients with Huntington's disease (N=4,007), which should reflect the underlying population distribution of the HTT CAG allele.
Clinical Cohort 3
Patients with a clinical diagnosis of major depression were recruited from consecutive admissions to the inpatient unit of the Department of Psychiatry and Psychotherapy of the University of Bonn, Germany. DSM-IV diagnosis was made by psychiatrists or psychologists applying a consensus best-estimate procedure based on all available information, including a structured interview (the SCID), medical records, and family informants when available. All patients (N=601) included in the study were of Caucasian ancestry, based on self-reported information about grandparents. The mean age at interview was 47.4 years (SD=13.8), and the mean age at onset of major depression, defined as when DSM-IV criteria were fulfilled for the first time, was 36.4 years (SD=13.2). The cohort was 63.7% women.
The comparison sample (N=1,339) was recruited within the framework of the German National Research Project in the region of Bonn to serve as a comparison sample for genetic studies of several neuropsychiatric phenotypes. Population-based recruitment was performed in collaboration with the local census bureau. Participants were evaluated by an experienced psychiatrist and screened for neurological and psychiatric disorders with a structured questionnaire. The mean age at interview was 47.8 years (SD=15.3), and the cohort was 52.2% women.
All participants provided written informed consent. All participants also consented to transfer of DNA and phenotypic data in an anonymized fashion to other research groups. Protocols and procedures were approved by the local ethics committees.
Molecular Methods
HTT CAG repeat allele sizes were determined by polymerase chain reaction (PCR) using an assay that does not include the adjacent proline (CCG) repeat and that is a modification of the method reported by Warner and colleagues (
29).
HTT CAG repeat sizes were determined by using a fluorescently labeled/tailed primer pair, CAG1-6FAM-ATGAAGGCCTTCGAGTCCCTCAAGTCCTTC and CAG2-GGCGGTGGCGGCTGTTGCTGCTGCTGCTGC. The PCR conditions were as follows: 80 ng of genomic DNA, 1.25 mmol MgCl
2, 1X Buffer II (Applied Biosystems), 0.5 U AmpliTaq Gold (Applied Biosystems), 0.125 μmol each primer, 0.25 mmol deoxyribonucleotide triphosphate, and 12% dimethyl sulfoxide in a 10 μL reaction. The cycling conditions for the reaction were as follows: initial denaturation at 94°C for 4 minutes, followed by 35 cycles of denaturation at 94°C for 30 seconds, annealing at 65°C for 30 seconds, extension at 72°C for 45 seconds, and a final extension of 10 minutes at 72°C. The PCR products were run on an Applied Biosystems 3730XL DNA Analyzer with GeneScan500 LIZ (Applied Biosystems) as the internal size standard. The data were analyzed with GeneMapper v3.7 (Applied Biosystems), and the sizes were converted to CAG repeat number based on comparison to known CAG repeat number standards.
Analysis
We defined
HTT risk alleles (completely or incompletely penetrant) a priori as those with a length ≥36, the threshold at which Huntington's disease risk appears to emerge (
3). Fisher's exact test was used to compare frequency of
HTT risk alleles among case and comparison subjects.
Power for an alpha level of 0.05 was estimated by simulation using the PLINK software tool set (
30), assuming a genotype relative risk of 6 and a minor allele frequency of 0.001, using Fisher's exact test. Power to detect association under these conditions was 23.5%. Stata 10.0 (Stata Corp., College Station, Tex.) was used for all analyses.
Results
In cohort 1, two of 68 chromosomes from patients with major depression had
HTT CAG repeat allele sizes ≥36 (lengths of 36 and 37), compared with none of 148 chromosomes from participants in the comparison group (Fisher's exact test, p=0.098). In cohort 2, a total of four alleles of 36 repeats or greater were observed (lengths of 36, 37, 38, and 42) among 2,986 chromosomes from depressed subjects, compared with none of 4,007 control chromosomes (Fisher's exact test p=0.006).
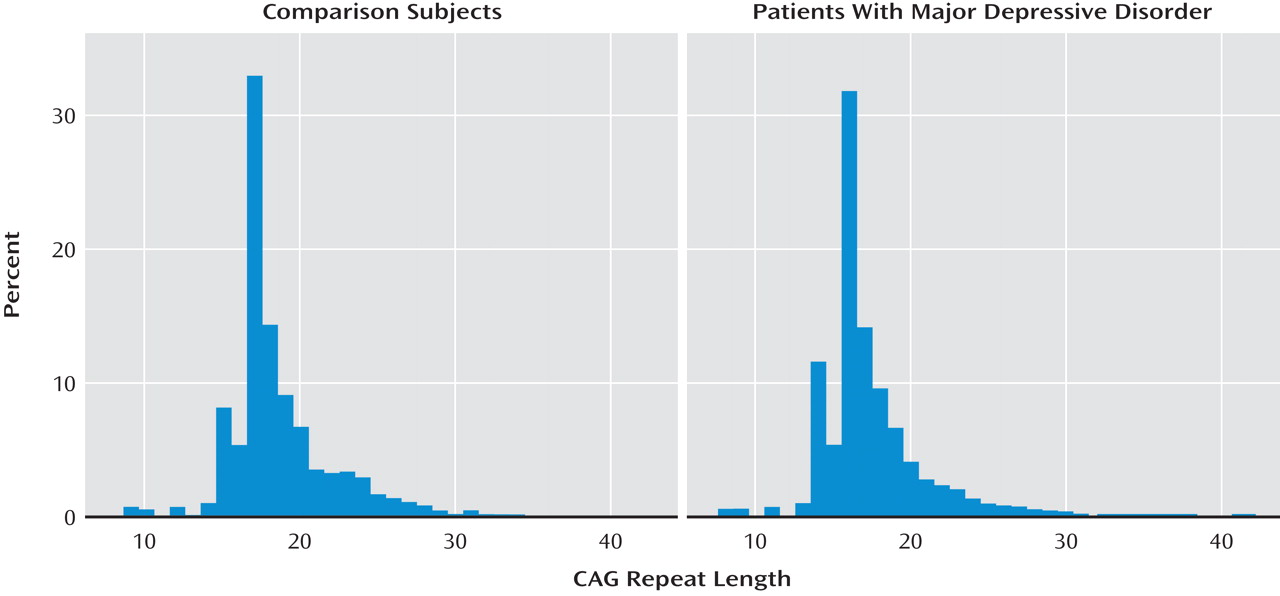
Figure 1 shows the distribution of
HTT allele lengths among patients with major depression and healthy comparison subjects in this second cohort.
In the replication cohort (cohort 3), one expanded allele (N=38) was identified among 1,202 chromosomes from patients with major depression, and none of 2,678 chromosomes from comparison subjects (Fisher's exact test, p=0.31). For the three cohorts combined, we observed 7/4,256 expanded alleles compared with 0/6,833 among comparison chromosomes (Fisher's exact test, p=0.001).
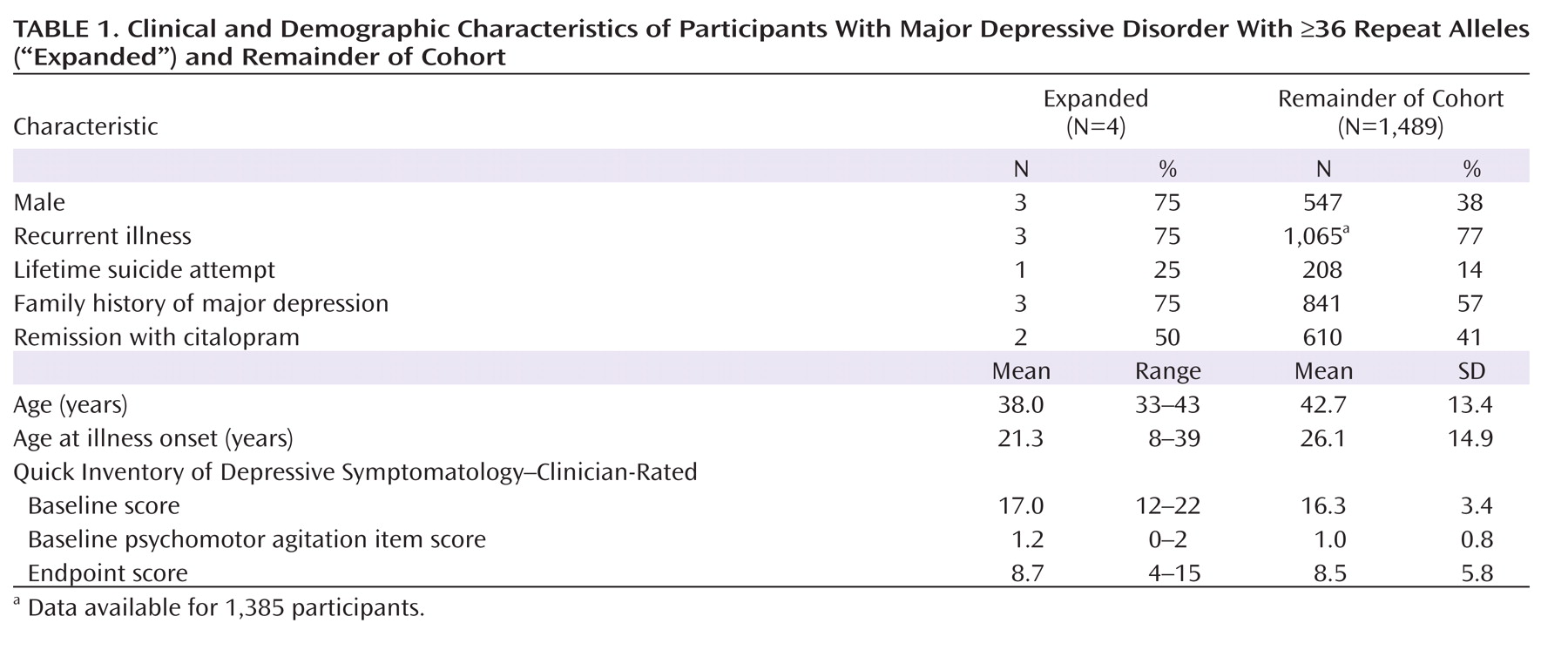
Table 1 compares demographic and clinical features for four participants with allele lengths ≥36 with the remainder of cohort 2. No pattern of sociodemographic or clinical features was apparent. Of note, the one participant with an allele length of 42 (i.e., in the fully penetrant Huntington's disease range) showed clinical improvement with citalopram treatment. Structural neuroimaging in 84 participants from cohort 1 (34 with major depression, 50 comparison subjects) indicated that caudate volumes for two individuals with major depression with expanded alleles and for the major depression cohort as a whole were not significantly smaller in volume relative to those of comparison subjects. (The normalized caudate volumes were 0.60 [SD=0.01] for each group; see the data supplement that accompanies the online edition of this article.)
Discussion
We identified seven expanded CAG repeats among patients with major depression and none among healthy comparison subjects after examining a total of 11,089 chromosomes. While the distribution of HTT CAG allele lengths in unselected populations has not been well established, our results suggest that low-end Huntington's disease-associated alleles are overrepresented among individuals diagnosed with major depression. Specifically, our results would estimate that ∼7/2,128, or 3.3 in 1,000, individuals diagnosed with major depression carry an expanded HTT CAG allele (95% confidence interval=0.9–5.7).
The most straightforward interpretation of our results would suggest that all of these individuals will ultimately develop Huntington's disease with time and that the depressive symptoms simply represent a prodromal presentation, as other studies have suggested (
5,
6). Indeed, all of the patients with major depression reported on here are below the predicted age at onset of Huntington's disease based on repeat number (
31,
32). This interpretation would suggest a parallel with the 22q11 deletion associated with velocardiofacial syndrome but also seen in clinical samples of patients previously diagnosed with apparent schizophrenia (
18,
19). While one could hypothesize some enrichment of expanded Huntington's disease alleles among individuals with depressive or irritable symptoms, our results indicate a greater-than-expected prevalence among individuals diagnosed with major depression. They would underscore the importance of inquiring about family history of movement disorder and, where indicated, performing a careful neurologic examination in individuals with depressive symptoms.
Alternatively, our results may shed light on the phenotype corresponding to incompletely penetrant alleles. Molecular studies indicate that the CAG repeat length affects mitochondrial ADP phosphorylation (
33), even for CAG repeat lengths in the intermediate range. However, no clinical phenotype has previously been described for these low-end
HTT alleles, except when they cause the overt clinical symptoms of Huntington's disease. Our results would also be consistent with a model in which incompletely penetrant alleles are associated with risk for presenting with depressive symptoms even if they never lead to the movement disorder characteristic of Huntington's disease. This hypothesis may merit investigation in presymptomatic Huntington's disease cohorts. The mean age of individuals with expanded CAG repeats here is more than two decades before the predicted onset age of motor symptoms based on allele length, and thus earlier than psychiatric symptoms are typically reported (
31,
32). Notably, data from a Huntington's disease mouse model suggest that the antidepressant fluoxetine improves depression-like behaviors associated with characteristic Huntington's disease pathology (
34), which might explain the observed benefit of antidepressant treatment in some of the cases identified in our study.
Limitations of this study include the absence of a neurologic examination of sufficient scope to rule out Huntington's disease motor symptoms in cohort 2 and the absence of systematic collection of family history of neurologic diseases in any of the three cohorts. Therefore, we cannot exclude the possibility of unrecognized Huntington's disease, particularly in cohort 2, although it is important to underscore that for all participants in the depression group, the primary diagnosis was major depressive disorder, and thus individuals with known or suspected Huntington's disease would not have been enrolled. As DNA was provided to us in an anonymized fashion and recontact with participants was prohibited by the institutional review board, it is impossible to obtain further neurologic examination or longitudinal follow-up data to determine which of these individuals will develop Huntington's disease motor symptoms. With this limitation in mind, more detailed clinical characterization and longitudinal observations of individuals with HTT CAG alleles in the incompletely penetrant range would be of great value.
We also caution that the power to detect association is limited (∼23.5%) even with multiple large cohorts, because the risk variant is rare. For a genome-wide study, the risk of spurious association (i.e., a low posterior probability) would be great under these conditions, as both prior probability for any given locus and power are low. Conversely, in the present study, where only one locus was tested based on a high prior probability of association, the posterior probability would be substantially greater despite modest power. Still, analysis of additional cohorts of patients with major depression and comparison subjects will be important to estimate the risk allele frequency with greater precision.
These results add to a growing body of literature suggesting that major depression is a heterogeneous illness whose presentation overlaps with neurodegenerative disorders. That is, some individuals presenting with a major depressive episode may be prodromal for a neurodegenerative illness. For three of the paradigmatic neurodegenerative disorders—Alzheimer's disease, Parkinson's disease, and Huntington's disease—depressive symptoms may precede other typical disease symptoms, in some cases by a decade or more (
5,
6,
35,
36). In Parkinson's disease, there is evidence of shared disease liability with depression: first-degree relatives of individuals with Parkinson's disease are at elevated risk for depressive and anxiety disorders (
37). Carriers of risk genes for other neurologic disorders believed to affect the basal ganglia, such as
DYT1 in idiopathic torsion dystonia, likewise have an elevated risk for recurrent major depression (
38). Further studies examining disease risk genes for neurodegenerative disorders, particularly those affecting the basal ganglia, among cohorts with major depression may help to elucidate the pathophysiology underlying depressive symptoms in general. These studies may also help to clarify the utility of further examination for neurologic disorders in patients presenting with a major depressive episode.