Autism spectrum disorder (ASD) comprises a group of developmental disorders characterized by impairments in social interaction and communication and stereotyped interests and behaviors (
1). Consistent increments in the observed prevalence of ASD are acknowledged as a pressing public health concern, with up to 2% of children developing ASD (
1). The importance of genetic causes has long been recognized, and substantial advances have been made in identifying genetic contributions, including rare de novo variants strongly related to ASD and common polymorphisms that provide weaker contributions to risk (
2). However, recent studies (e.g.,
3) have highlighted the potential for nongenetic or shared environmental factors to play a significant role in the causes of ASD.
The processes that lead to ASD probably begin during fetal development, as signs can appear early in life. Poor fetal growth has been associated with a variety of cognitive and psychiatric problems in children (
4) that overlap with difficulties experienced in ASD and a range of adult mental disorders (
5). For example, poor fetal growth is related to speech and language deficits, internalizing and attention problems, social difficulties (
6), hyperactivity (
7), and learning disabilities (
8). In most studies with accurate recording of birth weight and IQ, birth weight up to 4,000 g has been associated with IQ even in same-sex sibling pairs (
9). Almost all studies of ASD report a substantial proportion of comorbid intellectual disability, although this varies across studies from approximately 15% to 60% (
1,
10).
Fetal growth is influenced by genetic and nongenetic factors (
4). A detailed understanding of how fetal growth and ASD are associated is likely to advance the search for causes of ASD with and without intellectual disability; however, key methodological problems challenge the existing evidence. First, many studies of the association between ASD and fetal growth have assessed birth weight, which is a rather crude measure of fetal growth. Second, other studies derive sex-specific birth weight for gestational age from live births, which yields a biased measure of birth weight for gestational age for preterm births because of selective survival (
11,
12). In Sweden, early ultrasound dating provides an intrauterine weight-based standard that is superior to the conventional birth weight for gestational age in yielding an unbiased measure and predicting perinatal morbidity and mortality (
13). Third, in studies of both birth weight and birth weight for gestational age, most have dichotomized exposure into normal versus low birth weight (
14–
17), or appropriate for gestational age versus small for gestational age (
16,
18,
19), rather than examining associations between ASD and a continuous measure of fetal growth (i.e., the degree of deviance in growth above and below the norm in the general population from which case subjects derive). Fourth, study samples have been too small to provide enough case subjects in the extreme exposure categories (<2.5 kg or >4.5 kg) (e.g.,
14,
20). Fifth, studies rarely distinguish between ASD with and without intellectual disability in calculating risk. Since fetal growth is related to intellectual disability, the relationship between fetal growth and ASD may differ in individuals with and without intellectual disability. Finally, many studies have used selected subsets of children with ASD, such as children in psychiatric inpatient care (
16,
17). Thus, previous findings may not pertain to the broader population of children now identified on the autism spectrum.
We addressed these limitations by using the Stockholm Youth Cohort, a prospectively studied total population sample in which data from children with ASD were systematically ascertained (
21,
22). We examined fetal growth as a continuum in a sample of sufficient size to disentangle the relationships between fetal growth, gestational age, and ASD; to evaluate continua extremes; and to differentiate between effects with and without intellectual disability. We hypothesized that deviance from the mean in growth (either below or above) would be associated with greater risk of ASD.
Method
Design and Study Population
The research ethics committee at Karolinska Institute granted approval for the study. We conducted a matched nested case-control study within the Stockholm Youth Cohort (
22), which includes all children ages 0–17 who resided in Stockholm County between 2001 and 2007 (N=589,114). The Stockholm Youth Cohort population was identified using the Swedish Registry of the Total Population (
http://www.socialstyrelsen.se/statistics;
23), and prospective data from probands and their first-degree relatives were collected using records linked with Swedish national and regional health and administrative registries (
22). Exclusion criteria and characteristics of the sample included in our study are listed in Figure 1 of the data supplement that accompanies the online edition of this article. First, we excluded those who had not resided in Sweden or Stockholm County for at least 4 years (N=144,187). This enabled us to exclude children too young to have a diagnosis and ensured that migrant children with ASD had enough time to undergo assessment and diagnosis. From the remaining children, we excluded all adoptees (N=5,636). This resulted in 439,291 children and adolescents. We then excluded children who were not born in Sweden, whose obstetric information was not available (mainly those who were born abroad), and twins. These criteria excluded approximately 10% of the remaining individuals. Data on other relevant covariates were missing from 4%, leaving 4,283 children with ASD (1,755 with intellectual disability and 2,528 without) and 36,588 age- and sex-matched healthy comparison subjects who were randomly selected from the control population in the Stockholm Youth Cohort. Ten comparison subjects were originally matched per ASD child, but following the exclusions described above, a median of nine matched comparison subjects remained for each case subject.
Case Ascertainment
Data from children with ASD were ascertained using the unique identity numbers assigned to all Swedish residents and an exhaustive multisource linkage with registries covering all pathways of assessment or care of ASD in Stockholm County (
21–
24). In the Stockholm Youth Cohort, infants and preschool children are offered a structured assessment of social, motor, language, and cognitive development at centers whose health and surveillance program engages 99.8% of all preschool children (
22). Assessments are performed by trained nurses at regular intervals (1, 2, 6, 10–12, 18, 36, 48, and 60 months), at regular pediatrician examinations (2, 6, and 10–12 months), and at other intervals according to need or in case of developmental deviation (
22). Referrals for assessment of suspected ASD are commonly made by child health care centers and can also be requested by parents through family practitioners, schools, and other health or social care agencies.
Most autism-related services in Stockholm County (diagnosis, follow-up health, special education, and social care) use regional guidelines to assess developmental disorders like ASD and intellectual disability (
24). The guidelines recommend age-standardized neuropsychiatric cognitive testing and structured assessments of social, medical, and developmental history by parental report and by observation of the child in natural settings by specialist teams (
22). Structured follow-up is provided according to the presence or absence of comorbid intellectual disability (defined as IQ of 70 or less). Intellectual disability in the Stockholm Youth Cohort was ascertained as a comorbid diagnosis of ICD-9 codes 317–319 or ICD-10 codes F70–F79 and DSM-IV codes 317–319 and supplemented using the Habilitation Register, which categorizes service recipients as having ASD with or without intellectual disability. These categories were recently validated by a child psychiatrist and a neuropediatrician who investigated record-based evidence related to the diagnosis of ASD or intellectual disability according to ICD-10 and DSM-IV classification (
22).
Unlike in most population studies, our case subjects included those requiring health services and special education or social care. This is important because most children with ASD may not access health services after diagnosis, but receive input from social and educational agencies. Thus, we were likely to capture most children with an ASD diagnosis in Stockholm County with high validity (
21,
22). The 2007 prevalence of ASD in the Stockholm Youth Cohort was 114.8 per 10,000 (95% confidence interval [CI]=111.7–118.0) (
22), which is almost identical to other population prevalences. We were able to differentiate between ASD with and without intellectual disability, following the service delivery models in Sweden, which is an informative subclassification strategy (
25). We were unable to study ASD according to the subclasses used in current classification systems because this information was not consistently available in the registers.
Exposure Variables
Prospectively recorded information on birth weight and gestational age was retrieved from the Swedish Medical Birth Register (
26). Since 1990, approximately 95% of Swedish women have received ultrasound screening in the second trimester; ultrasound measurements of fetal size determine the gestational age of the fetus and thus gestational age at birth and birth weight for gestational age. The pregnancies without ultrasound data were dated from the first day of last menstruation. We calculated z scores of deviance in fetal growth from the reference curve for estimated fetal weight, because an intrauterine weight-based standard is superior to the conventional birth weight for gestational age standard in predicting perinatal morbidity and mortality (
12). These z score groups were stratified according to conventions of standard deviations as narrowly as the data would allow at the extremes of distributions. Gestational age was stratified to represent recognized clinical categories of pre- and postterm birth.
Potential Confounders
Maternal and paternal psychiatric history, parental age at birth, and socioeconomic status predict elevated risk of ASD (
27–
30). Parental psychiatric history was ascertained from the Stockholm County Council Adult Mental Health Service Register (all adult psychiatric outpatient care in Stockholm County since 1997) (
27,
31) and the National Patient Register (discharge diagnoses for inpatient psychiatric episodes since 1973) (
23). Parental psychiatric history included any psychiatric care in neither, one, or both parents. We recently detailed the relationships between psychotic diagnoses in parents and ASD risk in their offspring in three cohorts (
27). The Swedish Multi-Generation Register was used to identify biological first-degree relatives and parental age at birth and parental countries of origin (coded as Sweden or other). The Longitudinal Register for Labor Market Research and Insurance (
23) provided prospectively recorded information on socioeconomic status, including income, occupational class, and parental level of educational. Individual disposable family income for biological mothers was coded into quintiles in relation to year of birth. Parental education has been found not to be associated with ASD risk and therefore it was not assessed as a covariate (
30). We retrieved data from the Swedish Medical Birth Register (
26) on gestational diabetes and hypertension and on congenital malformations in the child, which are known to be associated with abnormal fetal growth (
32–
34).
Statistical Analysis
In the nested case-control sample, we conducted conditional logistic regression to calculate crude and adjusted odds ratios with 95% confidence intervals as estimates of relative risk of ASD overall and ASD with and without intellectual disability in relation to perinatal characteristics. These were adjusted for parental age, country of birth, occupational class, household income, family psychiatric history, gestational diabetes and hypertension, and congenital malformations in the child. Although z scores take gestational age into account, this variable remains a potential confounder of the association of ASD with fetal growth because reduced fetal growth increases the risk of spontaneous preterm birth, and because risk factors for impaired fetal growth and preterm birth overlap (
4,
11,
12). To account for this, and to assess the independence of the observed associations, we examined risk of ASD according to a cross-classification of fetal growth and gestational age. We performed statistical tests of the interaction between gestational age and fetal growth z scores using the synergy index (
35). Analyses were conducted using SPSS, version 20 (IBM, Armonk, N.Y.).
Results
Table 1 summarizes the demographic characteristics of the 4,283 children with ASD and the 36,588 matched comparison subjects (data separating ASD with and without intellectual disabilities are available in the online data supplement).
In large samples, small absolute differences can be statistically significant. In these data, the following differences were of large magnitude and significant: parents of children with ASD were more likely to have experienced psychiatric admission than comparison subjects (18.7% and 11.3%, respectively), and children with ASD were more likely than comparison subjects to have congenital malformation. There were small significant differences between children with ASD and comparison subjects on lower occupational class and lower income (see
22,
30). Conditional logistic regression models (
Table 2 and
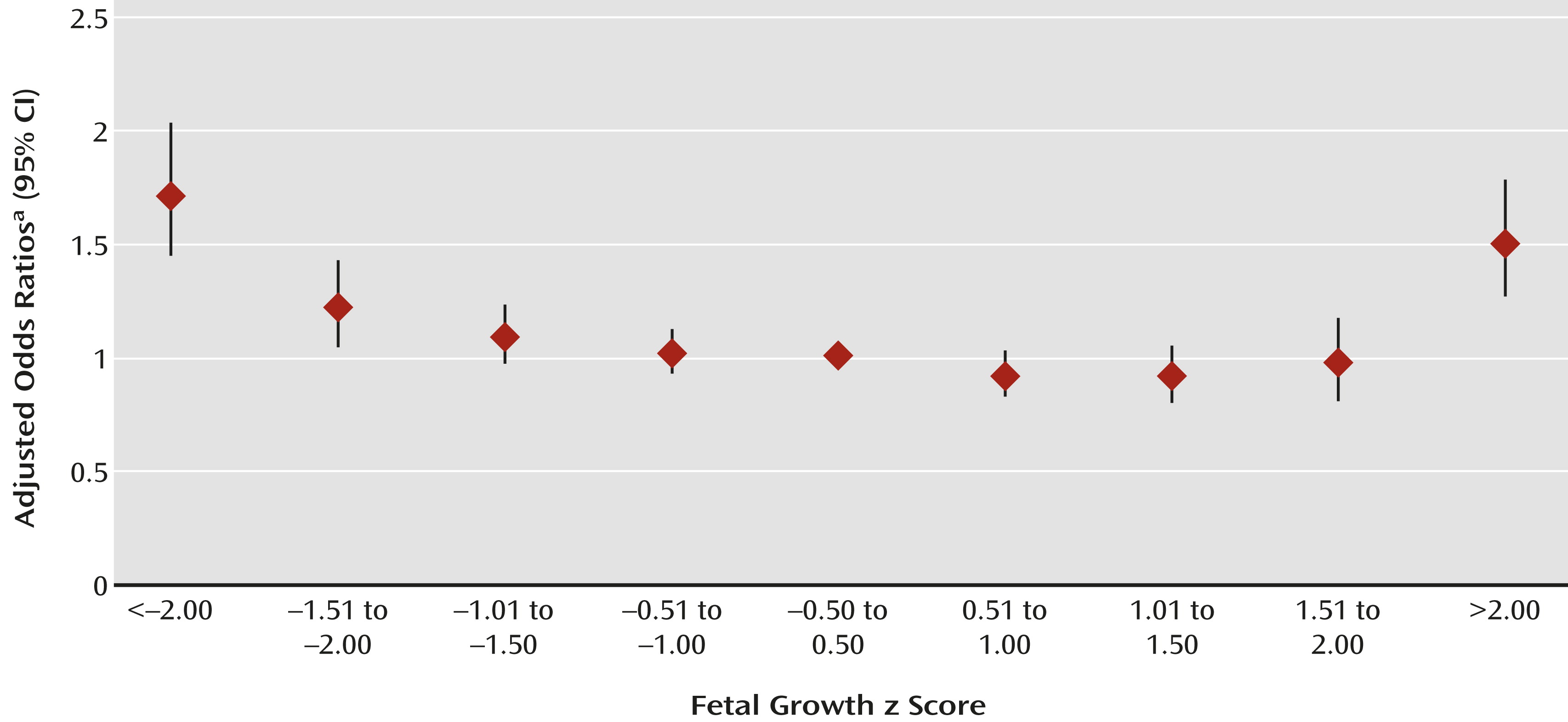
Figure 1) indicated greater ASD risk with fetal growth z scores 1.50 standard deviations below and >2.00 standard deviations above the mean for gestational age. Relative risk rose with increasing deviation below the mean; it was greatest for <2.00 standard deviations below the mean (odds ratio=1.8, 95% CI=1.6–2.2). Above the mean, ASD risk increased only at the extreme (>2.00 standard deviations above the mean: odds ratio=1.6, 95% CI=1.3–1.8). These relationships followed similar patterns when children with ASD were stratified by sex (see tables in the online data supplement); the interaction between sex and fetal growth was not significant. These relationships were observed for children with ASD with and without comorbid intellectual disability (
Table 3). Fetal growth below the mean was more strongly associated with ASD with intellectual disability than without (p<0.004). Parental age, psychiatric history, socioeconomic status, country of birth, congenital malformation in the child, and gestational diabetes and hypertension had only modest confounding effects on the relationship between fetal growth and ASD with and without intellectual disability (
Tables 2 and
3).
Table 4 summarizes the relationships between fetal growth, gestational age, and ASD risk. Children born small or large for gestational age were at greater ASD risk across all gestational age strata. Regardless of fetal growth, prematurity was positively associated with elevated risk. We observed an interaction between fetal growth and gestational age, such that growth restriction conferred particularly greater risk in children born postterm (synergy index 3.0, 95% CI=1.3–6.6). Patterns of risk were similar for ASD with intellectual disability and ASD without intellectual disability, but we observed stronger effects in the ASD with intellectual disability group (data available on request).
Discussion
To our knowledge, this is the first large prospective population-based study to describe the association between deviance in fetal growth and ASD in children with and without comorbid intellectual disability. There were four main findings. First, poor fetal growth was related to greater risk of ASD, which was most evident at the extreme of poor fetal growth (i.e., <2.00 standard deviations below the mean). Second, deviance in growth at the opposite distribution extreme (>2.00 standard deviations above the mean) was also related to increased risk. Third, although patterns were similar overall for ASD with and without intellectual disability, the association with poor fetal growth was stronger for ASD with intellectual disability. Fourth, preterm birth was associated with greater ASD risk independently of poor fetal growth, and vice versa.
Although the results of some population studies were inconclusive (
16,
18,
19), our findings are broadly consistent with previous studies that used birth weight as a proxy for fetal growth and reported significant associations between increased ASD risk and low birth weight (i.e., <2.5 kg as defined by the World Health Organization) (
14,
15,
17). A recent study of a low birth weight cohort (
36) also reported a significant positive association between ASD and birth weight below 2 kg, and a contemporary meta-analysis reported a significant association with low birth weight (relative risk=1.63, 95% CI=1.19–2.33) and with small-for-gestational-age infants (relative risk=1.35, 95% CI=1.14–1.61) (
37). To our knowledge, the association between ASD and fetal growth 2.00 standard deviations above the mean has not been previously reported. This association reached significance in all models and was robust in children with ASD with and without intellectual disability. Smaller population studies (
16–
18,
20,
38,
39) and meta-analyses (
37) reported either no risk or nonsignificant excess risk above 4.5 or 5 kg or in large-for-gestational-age infants, which likely reflects insufficient statistical power in these studies to examine this association.
Prematurity was also associated with an elevated ASD risk. The independent effects of fetal growth and gestational age were consistent with those reported in previous studies (
16,
17,
20,
38). We also report significant interaction such that postmature, small-for-gestational-age infants showed particularly elevated ASD risks (
18). These infants may be dysmature, but postmaturity may also indicate abnormality of the fetal-placental unit, including placental insensitivity to processes that initiate delivery. In this case, infants exposed longest to factors associated with growth restriction, such as poor placental function, may be at greatest risk for poor outcome. Future research could usefully explore whether measures of fetal growth or gestational age are proxy exposures for such feto-placental dysfunction.
Most previous studies did not distinguish case subjects with or without intellectual disability. Eaton et al. (
18) reported increased risk only of ASD without intellectual disability in children with low birth weight, but not in small-for-gestational-age children, although the number of cases was small and the authors did not report associations for large for gestational age or high birth weight. Leonard et al. (
39) studied children with ASD within an intellectual disability cohort in Western Australia. In a relatively small number of children with comorbid intellectual disability and ASD, they reported no associations between birth weight or gestational age and the odds of ASD with intellectual disability. Infants born 24% above optimal birth weight were more likely to be diagnosed with ASD with intellectual disability, but the wide confidence interval (odds ratio=2.36, 95% CI=0.93–6.03) indicates limited statistical power. Our findings suggest that the degree to which an infant deviates from its optimal growth, at the extremes of higher or lower deviation from the mean, increases the risk of ASD with or without intellectual disability.
Strengths and Limitations
This study is distinguished by particularly strong features. It was truly a population-based study rather than an inpatient or hospital discharge sample. The study sample contained nearly complete prenatal and outcome data with a very large sample size, and we were able to adjust for a comprehensive range of relevant confounders.
There are some important limitations to consider. Apart from gestational diabetes and hypertension, we did not adjust for intranatal or postneonatal events or maternal and neonatal morbidity. Swedish data suggest that maternal and neonatal morbidity are likely to be risk mediators (
24), and therefore inclusion may bias estimates. We did not measure a number of maternal factors that may be involved in poor fetal growth (e.g., alcohol and substance misuse, exposure to prescribed medication, or nutritional influences) (
40). Similarly, maternal prepregnancy obesity and gestational weight gain may also be involved in the development of macrosomia (birth weight >4 kg or >90th percentile) (
41). A measure of head circumference at birth was available, but we were unable to adjust for this because it was not measured consistently across the study period. We adjusted for psychiatric illness in mothers and fathers, but cannot account for a range of family-level effects that may be confounders (
42). Dodds et al. (
20) suggested that children with a positive family history of mental illness, including autism, may be predisposed both to suboptimal obstetric factors and to ASD. Nonetheless, a recent large population study (
43) using a cotwin-control design reported that in twin pairs discordant for birth weight, twins with lower birth weight were significantly more likely to score above the cutoff for likely clinical disorder on a scale predictive of ASD. This twin-control design reduces potential confounding that might influence the association between birth weight and ASD, and the results imply that nongenetic influences associated with birth weight may indeed be important antecedents in the development of ASD (
42). Finally, it would have been interesting to compare our findings with a well-defined control group of children who had intellectual disability but not ASD, but this was not available in the present study.
Deviance in fetal somatic growth may co-occur with abnormalities in the trajectory of fetal brain development in ways that are specifically, or nonspecifically, linked to ASD. Like the association between fetal growth restriction and lower IQ in the general population (
9,
42,
44), fetal growth restriction may favor the development of ASD with intellectual disability and could provide clues to the mechanisms specific to this aspect of illness. There remains a need to investigate how and why fetal growth that deviates from the population average is related to greater risk of ASD in children.
Research into the control of fetal somatic and brain growth increasingly implicates placental function and early placental programming of infant development (
4). In adult metabolic and cardiovascular risk, examination of the ways in which environmental and genetic influences on development interact has shown that environmental perturbations during fetal development have a lifelong effect on the gene-informed trajectory of endocrine and cardiovascular homeostatic systems (
4,
11,
45). Studies such as these are paving the way for novel preventive approaches to disease risk (
46).
Macrosomia was associated to a similar degree with higher risk of ASD with and without intellectual disability, and elevated risks persisted after adjusting for gestational diabetes. Genetic and epigenetic influences on macrosomia may have some specific effects on aspects of ASD pathophysiology that are not associated with intellectual functioning. Macrosomia and its important risk factors—gestational obesity and diabetes—are increasingly prevalent. Similarly, possible risk factors for reduced fetal growth, including maternal smoking, alcohol, and medication use, may be especially important given the greater likelihood that parents of children with ASD have a mental illness (
27). Further research is required to determine whether deviant fetal growth is a predictor or a cause of ASD, although these risk factors present an attractive target for primary prevention of idiopathic ASD because they are easy to identify and amenable to intervention (e.g., by preventing maternal obesity). Prematurity also increased the risk of ASD independently of fetal growth. Thus, although the majority of children with ASD were term infants, the improved survival of preterm infants suggests that this remains an important target for preventive intervention.
Overall, we believe that these findings provide important information for clinicians and parents. Associations of ASD risk with deviance in fetal growth suggest the possibility of early intervention to reduce poor developmental outcomes through careful antenatal monitoring as well as follow-up, screening, and management of infants who may be most at risk.
Conclusions
Deviance in fetal growth below the mean, particularly at its extremes, and preterm birth are strong independent risk factors for later ASD, especially with comorbid intellectual disability. By contrast, fetal growth at the extreme above the mean is linked with increased risk of ASD regardless of intellectual disability. These robust associations between ASD, fetal growth, and preterm birth provide further impetus for research assessing genetic as well as environmental risk in ASD. Understanding how genetic and environmental factors might influence fetal growth and its control during critical developmental periods may reveal important insights into the etiology of ASD.