This observed pattern of our current psychiatric science serves as a starting point for discussing the three central research paradigms for our field.
Paradigm 1: Populate the Major Levels and Sublevels With Validated Risk Factors
Given the dispersion of risk factors for psychiatric illness, we need to identify as wide a range of etiological factors as possible, with the only methodological concern being confidence in causal attribution. The ideal conceptual framework for this approach is the interventionist model of causality (
6,
7). This approach seeks “difference makers”—where making a change in a putative risk factor alters the probability of psychiatric illness. An attractive feature of this model is its lack of philosophical baggage. As long as empirical data show that variation in the risk factor is directly associated with variation in risk for the disorder (and not the result of confounding factors), this approach can accommodate phenomena occurring at supra-individual levels (e.g., increasing social capital in neighborhoods through community interventions reduces risk for drug abuse [level E3]), within individual factors best understood at a psychological level (e.g., increasing levels of self-esteem through therapy protects against depression [level P3]), and various levels best understood using neuroscience or molecular methods (e.g., increased sensitivity to adverse stimuli in fear circuits predicts anxiety disorders [level B3], or variation at specific alleles predicts risk for schizophrenia [level B1]). In particular, this model of difference makers is insensitive to the mind-body problem and captures the fundamentally practical goals of psychiatric science. We want to intervene on our risk factors to reduce rates of psychiatric disorders. Clinicians themselves want to be difference makers, preventing the progression of disease pathways.
The problems of causal inference in etiological psychiatric research are often challenging. Outside of animal models, randomized experiments are often impractical or unethical. The organization of biology gives us a few privileged areas (e.g., genomic DNA variation causes phenotypes and not the other way around), but otherwise, discriminating causal effects from the impact of confounders can be difficult. We have two basic approaches to this problem: natural experiments—such as a co-twin control design—and statistical methods—such as propensity matching (
8,
9,
10). We pay insufficient attention to this critical question, although in the past decade our field has seen creative uses of natural experiments to answer key causal questions (
11–
13).
In this first paradigm, we need not focus on the specifics of the disease pathway. More formally, the interventionist model makes no assumptions about the nature of the mediational mechanisms between the risk factor and the disorder.
Why are there so many diverse research traditions examining the origins of psychiatric illness? Part of the driving force behind the pluralism in our field is divergent epistemic values. That is, we do not agree on what are the most important things to know about psychiatric illness. There are many ways to try to judge the adequacy of different scientific explanations (e.g., disordered functioning in hippocampal GABA cells increases risk for schizophrenia, or home foreclosure increases risk for depression) and a long series of proposed criteria (simplicity, completeness, generalizability, etc.). However, ultimately our preferred explanations reflect values about what we want to know. In our field, we have a deep debate about the relative values of explanations grounded in the biological sciences and in the realms of the mental and the social. This debate reflects our unusual professional identity, sitting at the crossroads of biomedicine, the social sciences, and the humanities. We also have more pragmatic debates. Do we want to maximize our predictive power or seek basic biological understanding? Taking genetics as an example, predictive power is maximized at level B4 (aggregate genetic effects). Biological understanding, however, can be much better accomplished at level B1 (molecular genetics), where individual genetic variants or networks of variants identify specific pathophysiological pathways. While some might see the wide range of our research traditions as a liability—and surely the problems we deal with can be bewilderingly complex—this diversity can also be a strength if accompanied by sufficient scientific rigor.
We can make substantial scientific progress in clarifying the etiology of psychiatric illness by working at diverse single levels. As argued by Chang (
1), there have historically been important benefits to scientific fields in supporting multiple complementary research programs. Many fields would have suffered had there been a premature closure and focus on a single explanatory approach. The sources of scientific progress are difficult to predict. Consider the derision with which the theories of continental drift (
14) or prion disease (
15) were initially greeted. Disagreements can stimulate mutual criticisms, and responding to criticism often improves theories. Particular value can arise from having diverse research perspectives with distinctive methodologies focused on the same problem.
Here I need to reach ahead and make a crucial connection between this paradigm and paradigm 3 (bringing things back to the mental). Many of our risk factors can be easily articulated in third-person space—genetic markers, degree of urbanization, scores on a personality test. But some risk variables have been first conceptualized as mental processes understood from a first-person perspective. Some still require assessment using empathic approaches, or “imaginative understanding.” Take the development of the cognitive theory of depression by Beck (
16). This theory originated in listening carefully to dream sequences from depressed patients and hearing repeated themes of helplessness. Consider the concept of humiliation after stressful life events. The optimal way to rate humiliation is to have humans listening to audiotapes rating “what it was like to have been that person in that situation.” This is what Dennett terms heterophenomenology—a method of neutrally assessing first-person experiences without having to confront the hard problems of consciousness (
17, pp. 66–98). A central addition to Dennett’s approach—and critical to paradigm 1—is to verify the results empirically, showing them as legitimate risk factors. Beck’s theory translated into scales that predict and a therapy that treats depression (
16). Under blinded conditions, humiliation powerfully predicts onset of depression (
18,
19). We cannot fully populate our “causal space” for psychiatric illness, as outlined in paradigm 1, without our imaginative understanding.
What does this multilevel, empirically based pluralism tell us about the value of reductionism? To answer requires a distinction between global and local reductionism. Global reductionism argues that biological variables are inherently superior as an explanatory level for psychiatric disorders. A corollary is that we should focus our research time and resources solely on biological-level variables. As an example of local reductionism, consider the major effort now under way, driven by technological advances in genotyping and sequencing coupled with large-scale collaborative clinical projects, to move psychiatric genetic findings from the aggregate (B4) to the molecular level (B1). That this represents a potentially important advance—one that can move our etiological models from latent genetic factors to individual molecular pathways, which can then open up new avenues for pathophysiological understanding leading to potential novel drug targets—is incontrovertible. Another local reductionist agenda uses powerful epidemiological data sets and statistical/conceptual methods from analytic sociology (
20) to transition from latent estimates of environmental effects (such as those obtained from twin or adoption studies) to specifying, in explicit social science terms (such as exposure to social deprivation or peer deviance), the mechanisms of social transmission of risk of illness (
21,
22). We can, within individual domains, recognize that the power of moving from global latent constructs to detailed mechanistic processes provides greater scientific insight and more capacity for specific interventions, without having to make any overall assumptions about the relative superiority of biological, psychological, or epidemiological variables. Thus, empirically based pluralism would strongly support local reductionist research agendas but argue against global reductionism as an approach to understanding the etiology of psychiatric illness.
Paradigm 2: The Clarification of Causal Mechanisms
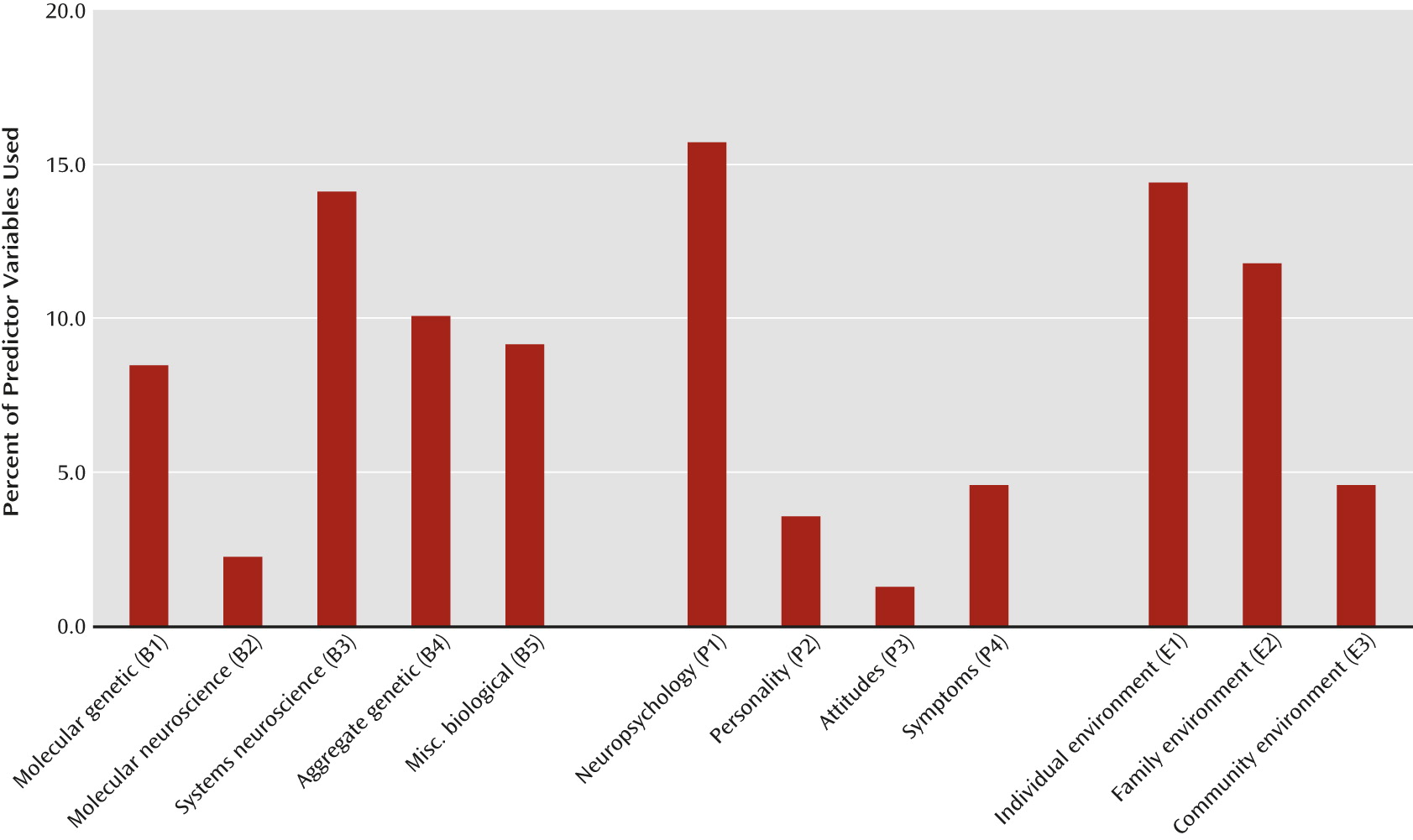
A focus on a single level of etiological explanation remains the predominant model in our research today, accounting for 69% of the studies reviewed. Because every major psychiatric disorder that has been the subject of careful research has risk factors across multiple levels (
5), to achieve a fuller understanding of causal processes, we need to develop multilevel mechanistic accounts of disease etiology (
4). It is inevitable that risk factors to be studied would include those understood initially at environmental, psychological, and biological levels.
Just as I recommended Woodward’s interventionist model as a guide for our first paradigm, the “mechanistic movement” in the philosophy of biology can provide important guidance for our second paradigm (
23–
29; for succinct recent reviews, see references
20,
30). Traditional philosophy of science, with a dominant physics orientation, saw the task of science as discovering deep underlying laws, à la Newton. Philosophers of biology have moved away from this conceptualization. To understand a biological system is rarely to see it as a manifestation of a few simple underlying laws. Rather, it is to figure out “how the damn thing works.”
What does “mechanistic” refer to in this context? Most simply, it involves seeing how individual “parts” work together to achieve an outcome that none could produce on its own. Consider a house heating system. It might have a furnace, an oil tank, pipes, wires, and a thermostat. Together they keep the house within a set temperature range. From a mechanistic perspective, an adequate scientific model would depict how the parts work together as a whole. Simply clarifying the operation of individual components of the mechanism would not be explanatory in the right way.
A mechanistic approach to the problems of psychiatry has several appeals. First, it is a natural next step after populating our risk factor domains. While this first stage involves a focus on identifying risks and trying to verify that the relationship is causal, a more demanding mechanistic approach now requires filling in the missing steps from the risk factors to the disorder. Understanding mechanisms requires a reductionist descent into the nitty-gritty of the world to figure out how things actually work. But in neurobiological systems, events always sit within contexts, and causal processes are typically multilayered. Mechanistic explanations therefore require the integration of multiple organizational levels. Second, it is essentially pragmatic in approach and avoids the ideological extremes of hard reduction on the one hand and mysterian emergence on the other. The decomposition of a problem required by a mechanistic explanation (“let’s start by getting all the parts clarified”) is largely a reductionist task. But the recognition that parts and their interrelationships must be interrelated with one another into an appropriate whole requires a direct confrontation with the impact of multiple levels of organization. Bechtel, in particular, has written about how complex systems with causal loops and recursive cross-level interactions can, with patience and diligence, be decomposed and put back together again (
23,
24). Third, in outlining the mechanism of disease, it becomes possible to delineate the specific steps perturbed by particular risk factors and those that might be especially suitable for interventions aimed at prevention or treatment.
The mechanistic approach can help illuminate the central dilemma of psychiatric research—trying to match our causal mechanisms to our clinical disorders. We are at risk of committing a “lumping error,” in which we assume that several distinct syndromes are actually one, leading us to focus on a single underlying mechanism, when we should be seeking several distinct mechanisms. Alternatively, a “splitting error” would arise when several of our syndromes, thought to be clinically distinct, actually reflect the same underlying disease mechanism. On a broader level, our mechanistic turn will work well only if there is correspondence between our nosological system(s) and the underlying causal structure of psychiatric disorders.
What we need is more integrative and intuitive causal theories for the field of psychiatry (
31). As demonstrated by our literature review, we are constrained in our thinking about etiological processes. As Tenenbaum and colleagues write:
To learn the structure of causal relations between variables in a system, we need intuitive theories that generate hypotheses about alternative causal structures for that system. To learn such a theory itself, we need higher-order intuitive theories that generate hypotheses about theories at the next level down (
31, p. 304).
The most recent and powerful example of mechanistic explanation of complex biological processes is systems biology. Lander and Weinberg provide this succinct overview:
Twentieth century biology triumphed because of its focus on intensive analysis of the individual components of complex biological systems. The 21st century discipline will focus increasingly on the study of entire biological systems, by attempting to understand how component parts collaborate to create a whole (
32, p. 1781).
Noble adds to this as follows, noting the close collaboration between traditional reductionist models and the newer synthetic mechanistic approach, and pointing out that the critical difference is the bidirectional flow of causal processes:
An integrationist, using rigorous systems-level analysis, does not need or wish to deny the power of successful reduction. Indeed, he uses that power as part of his successful integration.… Integrative systems biology is just as rigorous and quantitative as reductionist molecular biology.… The only difference is that it accepts that causality goes from higher to lower levels as well as upwards (
33, pp. 66 and 77).
However, developing integrationist research programs presents both practical and conceptual challenges. Practically, the nature of specialization in scientific psychiatry makes it a challenge to develop excellent cross-disciplinary groups and to obtain the needed research samples and funding. Because the etiological pathways between and within levels can be complex and sometimes include dramatic nonlinear effects and causal loops, high levels of statistical sophistication are often needed.
A less often mentioned challenge of such multilevel research is that of incommensurability of key concepts across scientific communities (a point famously made by Kuhn [
34]). Quite literally, advocates for different levels within our field cannot always talk to one another because they have different vocabularies and understand key concepts in different ways (
2).
In Appendix 3 in the online
data supplement, I explore three examples of such incommensurability. Briefly, the term “depression” can be used to reflect current symptoms or a lifetime history of a neurobiological syndrome. Epidemiologists study objective features of the “environment” while twin researchers use the term “environment” to define all nongenetic factors that make individuals brought up in the same family similar or different. Key psychiatric constructs like anxiety and depression can be understood by different researchers as “states” or “traits.”
Our empirical review of current psychiatric etiological studies reveals two emerging cross-level proto-mechanistic research areas. The first is molecular and behavior genetics. This productive research field has gone from a largely descriptive approach (e.g., heritability calculations or odds ratios for individual SNP variants) to building increasingly complex models of illness including genetic and environmental risk factors and their interaction, correlation, and development together over time. The second vibrant area has been the increasingly close collaboration between neuropsychologists and systems neuroscientists, typically brain imagers. The bulk of this work now involves neuropsychological probes being given while individuals are undergoing functional MRI or positron emission tomography. More than any other current area in our field, this research collaboration is directly spanning the mind-brain divide. This collaborative venture also defines a “middle ground” for psychiatric explanation that can easily build on the ability to go “up” into other areas of psychological and environmental processes and “down” into molecular aspects of neuroscience. This level was posited by Murphy to constitute a particularly fertile level of explanation for psychiatric disorders (
35). In systems biology, researchers often try to move from top-down and bottom-up models to those that start in the “middle.” Noble captures this well:
The consensus is that it should be “middle-out,” meaning that we start modeling at the levels at which there are rich biological data and then reach up and down to other levels (
36, p. 102).
Perhaps the neuropsychology-systems neuroscience interface can function as the “middle-out” level for more detailed mechanistic explanations of psychiatric illness. We do not need to solve the mind-body problem to develop this important research interface, but we do have to be more sensitive to the problems of causal inference. Understanding whether and how subjectively reported psychological states are causal to, caused by, or share an identity relationship with the neuronal activity reflected in the blood-oxygen-level-dependent signal is a difficult problem that needs careful attention.
Paradigm 3: Tracing Causal Pathways Back Up Into the Mental
Once we have traced a mechanistic causal pathway for psychiatric illness down to a molecular neuroscience level, some in our field would argue that our task is done. We show that patients with disorder X have differences in brain structure compared with healthy subjects. We trace these differences to specific molecular and cellular pathways that we show to strikingly discriminate case and control subjects. We produce similar effects in model organisms. Then we declare victory and head home.
But this would be an incomplete scientific triumph. There is further integrative work to do involving tracing the causal pathways back up into the mind-brain system so that we can understand, first in neurobiological terms and then in mental first-person terms, how those disturbances cause the clinical symptoms and signs from which our patients suffer.
Psychiatry has long struggled with the question of how the mind works and becomes disordered in psychiatric illness. This goal should not be forgotten in the context of the exciting developments of molecular and systems neuroscience. Many times in the past, scientific developments have permitted humans to expand our experiences of the universe. The microscope and the telescope have allowed us to peer into previously unknown worlds. While the mind-brain problem poses subtler issues than optics, there are useful parallels. Neuroscience in general, and the functional analysis of the brain through neuropsychology in particular, can help us empathically understand aspects of human experience previously beyond our grasp.
The problem of linking the mental to more basic explanatory approaches in psychiatry is well illustrated by Frith, using examples that in our terms would be a direct explanation of psychiatric symptoms from levels B2 (molecular neuroscience) and B3 (systems neuroscience). He writes:
Certain causal explanations for schizophrenia symptoms are simply not admissible. For example, I think it is wrong to say “thought disorder is caused by supersensitive dopamine receptors” or “hallucinations occur when the right hemisphere speaks to the left hemisphere via a faulty corpus callosum” (
37, p. 27).
Why are these improper explanations? It might be true in an interventionist model that altering the sensitivity of the dopamine receptors increases the risk for thought disorder. But there are many steps from one to the other. We might think, “Well, this is like any other complex problem in medicine. Say we find that eating a high-cholesterol diet increases the risk for atherosclerosis. Does that mean we have explained atherosclerosis?” Clearly not. The problem in psychiatry is partly like that. We have to clarify the mediating physiological pathways from cholesterol ingestion to atherosclerosis involving processes such as inflammation, platelet aggregation, and so on. This is an approach of paradigm 2—clarifying the causal mechanism.
But in psychiatry, we have an additional question—the elephant in our room. We deal with symptoms of the mind. Stopping at third-person “explanations” of disease mechanisms leaves our project unfinished. We also want, and our patients deserve, understanding in a first-person framework. How is it that I have auditory hallucinations and delusions? Why did I develop these obsessions with this irrational fear of germs? The explanations need to be in psychological terms and not solely in biological terms.
Here we confront a central issue best articulated by Jaspers (
38) (after Wilhelm Dilthey), who suggested two qualitatively different kinds of knowledge: explanation, which utilizes natural sciences, objective and empirical methods; and understanding, which reflects our subjective, empathic appreciation of our patients’ experiences. Jaspers famously declared certain sets of psychiatric symptoms “off limits” to understanding, being literally “un-understandable” (
38).
This boundary is antiquated (
39). The intersection between systems neuroscience and neuropsychology has proved fruitful in developing “explanation-aided understanding” (
39). Take Kapur’s theory of ideas of reference arising from dysfunctions of the “dopamine salience” system (
40). He writes:
Dopamine mediates the conversion of the neural representation of an external stimulus from a neutral and cold bit of information into an attractive or aversive entity.… [T]he mesolimbic dopamine system is … a critical component in the “attribution of salience,” a process whereby events and thoughts come to grab attention, drive action, and influence goal-directed behavior.… (
40, p. 14).
Dopamine neurons encode motivational salience. In firing, they provide a signal: “This stimulus is important. Figure out what is going on!” What would happen if these dopamine salience neurons fired inappropriately? The result would be the incongruous intrusion into consciousness of meaning and significance. Whatever the person was seeing at the time—a yellow car on the road, a TV reporter reading the evening news—would suddenly be suffused with significance. It takes only a small step to imagine how an “un-understandable” idea of reference could be produced.
Using another combination of systems neuroscience and neuropsychology, Blakemore and Frith proposed a psychologically understandable model for the Schneiderian “made actions” (
41) based on the “feed forward model of motor control” (
42–
45). Similar efforts are under way using neuropsychology and systems neuroscience to explain verbal auditory hallucinations as misdirected inner speech and/or vivid memories generated internally in ways that the individual no longer recognizes as self-originating (
46).
We are dealing here with a different kind of knowledge from our mechanistic understanding with paradigm 2. Here we are seeking first-person understanding for our patients and enlarged powers of empathy for us. The story begins with individual causal risk factors, connected together through mechanistic cross-level processes that can then be re-expressed in comprehensible mental language. This is closing the circle. We begin our investigations with patients displaying symptoms. We categorize and study them. We clarify the nature of their underlying disorders. Our efforts are not complete until we have returned to where we started and can explain to our patients how their symptoms arose.