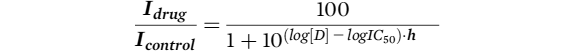Schizophrenia, a severe psychiatric disorder that typically emerges in adolescence and early adulthood, affects at least 24 million people worldwide (
1). Antipsychotic drugs, the cornerstone of treatment for patients with schizophrenia (
2,
3), block dopaminergic and serotonergic receptors (
2). These drugs, however, also block the voltage-gated K
+ channel Kv11.1 (
2,
4). Blockade of this channel has been thought to be an “antitarget,” as it increases the risk of QT prolongation and sudden cardiac death (
4). Other adverse effects of antipsychotic drugs include substantial weight gain, extrapyramidal symptoms and movement disorders, agranulocytosis, and many other less serious side effects, all of which are highly variable at the individual level (
5). There is similar variability in the therapeutic response to antipsychotics.
The Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) study was designed to identify differences in therapeutic benefit and liability for side effects between different antipsychotic drugs (
6). Of the nearly 1,500 patients with schizophrenia in the CATIE study, 74% discontinued their antipsychotic before the end of the trial at 18 months (
7). These patients had a variable response to all antipsychotics, both in terms of therapeutic benefits and side effects. Recent research by our group and others has sought to identify factors that contribute to the large variability in response to antipsychotics. We identified single-nucleotide polymorphisms (SNPs) in
KCNH2 that are associated with increased expression in human brain of a novel Kv11.1 isoform, Kv11.1-3.1, the expression of which is increased in schizophrenia brain (
8). Using data from the CATIE trial and a National Institute of Mental Health cohort, we found that patients who have
KCNH2 genotypes associated with up-regulated Kv11.1-3.1 expression in brain have an improved response to antipsychotic drug therapy in general (
9). However, whether there was any differential response between antipsychotic drugs was not determined. Another important factor in the variability of responses to pharmacotherapy is drug metabolism (
10). Recently, Almoguera et al. (
11) showed that patients who are slow drug metabolizers have a better response to treatment with risperidone compared with other antipsychotics. The variability in risperidone metabolism can be substantial. For example, in the CATIE study, the range of risperidone clearance varied 15-fold (range, 3.61–54.0 L/hour), and the clearance of paliperidone (9-hydroxyrisperidone) varied almost eightfold (range, 2.76–20.71 L/hour) (
12). However, what makes the differential treatment response in slow metabolizers so intriguing is that the principal metabolite of risperidone, paliperidone, has an affinity for dopamine D
2 receptors similar to that of the parent drug and therefore should produce a similar response if its therapeutic effects are primarily D
2-receptor mediated.
In this study, we investigated the hypothesis that the differential response to antipsychotic drugs seen in schizophrenia patients is related in part to differential block of Kv11.1 isoforms and drug metabolism. First, we found that risperidone, alone among the drugs tested, is a more potent blocker of Kv11.1-3.1 channels than of Kv11.1-1A channels. Surprisingly, its principal metabolite, paliperidone, as well as other atypical antipsychotics, including clozapine, olanzapine, and aripiprazole, showed no difference in the efficacy of block for the different Kv11.1 channel isoforms. Next, we found that patients enrolled in the CATIE study who have KCNH2 diplotypes associated with increased Kv11.1-3.1 expression and are also slow drug metabolizers (that is, have higher ratios of risperidone to paliperidone) show a marked symptomatic improvement when treated with risperidone compared with other drugs, while patients who are not slow metabolizers or who do not have Kv11.1-3.1-associated genotypes have a negative therapeutic response to risperidone. Our results highlight the potential for genotype-guided pharmacotherapy in the management of schizophrenia patients.
Method
Electrophysiology
Cell lines stably expressing Kv11.1-1A or Kv11.1-3.1 channels were maintained as previously described (
13). Patch clamp electrophysiology recordings were undertaken as previously described (
13; a summary is provided in the
data supplement that accompanies the online edition of this article).
Drug block was calculated as
Idrug / Icontrol and dose response curves were fitted with a modified Hill equation:
where
Idrug is current recorded in the presence of drug,
Icontrol is current recorded in control conditions,
D is the drug concentration,
h is the Hill coefficient, and
IC50 is the half maximal inhibitory concentration of D.
Data are presented as mean and standard error of the mean. Data were analyzed using one-tailed paired t tests and analysis of variance, followed by the Tukey t test for pairwise comparison. The significance threshold was set at 0.05.
Clinical Cohort
The clinical cohort consisted of patients randomly assigned to one of five antipsychotic medications during phase 1/1A (first drug assigned) of the CATIE trial. The details of the overall design for the CATIE study, genotyping of the
KCNH2 SNPs, and the participants’ demographic characteristics have been described previously (
6,
9).
Because of the outpatient and parallel design of the original CATIE study, information about compliance based on drug clearance is an important factor determining symptom change during the CATIE trial. Thus, we only analyzed treatment response from subjects of European ancestry for whom we had drug clearance and genotype data (N=362). We previously showed that drug clearance data substantially improve prediction of treatment response (
14). As an ancillary study to the CATIE trial, blood samples were drawn during study visits to measure antipsychotic drug concentrations. Data were collected on the amount of the last dose of medication, time the last dose was taken, and time the blood sample was drawn. This information was used with the drug concentration data to estimate drug clearance for each subject based on nonlinear mixed-effect modeling using NONMEM, version 5 (GloboMax, Ellicott City, Md.) (
12). A one-compartment linear model with first-order absorption (NONMEM ADVAN5) using the first-order estimation method was used to estimate drug clearance (
12).
We used estimated drug clearance instead of plasma concentrations because it is a dose-independent and time-independent measure, which allows for comparison of drug exposure across all subjects, as described in detail elsewhere (
14,
15).
For this analysis, we focused on three SNPs in
KCNH2—rs3800779 (SNP1), rs748693 (SNP2), and rs1036145 (SNP3)—which have been associated with increased expression of the novel Kv11.1-3.1 isoform in human postmortem brain samples (
8) and overall response to treatment in the CATIE trial (
9). Since the three SNPs were in moderate to strong linkage disequilibrium (see Table S1 in the online
data supplement), in order to reduce multiple testing and to gain statistical power for detecting association, we constructed three SNP diplotypes to be used for testing diplotype-by-risperidone interaction on the treatment response. Haplotype construction was performed and phased diplotype was assigned using the Phase program (
16). Details of genotyping and construction of diplotypes are provided in the
data supplement. Diplotype was grouped into three categories according to the number of minor alleles that a diplotype contains at SNP1 and SNP3, coded “0” for no minor allele of either SNP1 or SNP3, “1” for one or two copies of minor alleles, and “2” for three or four copies of minor alleles. The distribution of diplotypes in individuals with drug clearance data was consistent with the total European ancestry sample in phase 1/1A of the CATIE trial, suggesting minimal selection bias (see Table S2 in the
data supplement).
Clinical Data Analysis
In the CATIE sample, because all patients were receiving treatment and because the time and number of Positive and Negative Syndrome Scale (PANSS) evaluations in the study varied considerably among subjects, we treated the baseline PANSS rating as “before treatment” and the last rating as “after treatment” to test for genetic variant-by-risperidone treatment interaction on the treatment response. Since each subject had two measures in the analysis, we used a general linear mixed model to incorporate the relatedness between two observations within a subject (
9). We did not perform a separate analysis with only those subjects who completed the trial because that subset was too small (N=39).
We performed this analysis on all subjects for whom drug clearance data were available and for whom diplotypes were assigned with good confidence (N=362), and we controlled for potential covariates of sex, age, time on medication, and whether the patient completed the 18-month trial or discontinued medication before the end of 18 months and therefore switched to phase 2 of the trial.
Individuals who were on risperidone (N=88) had a mean estimated drug clearance rate of 20.62 L/hour (SD=10.73, range=3.61–40.05). Based on tertile distribution of the clearance data range, we classified individuals into three groups: slow (N=30), intermediate (N=29), and fast metabolism (N=29) groups.
The clinical response analysis consisted of a diplotype-by-treatment interaction, as previously described (
9). To test our specific hypothesis of a differential effect of diplotype on the antipsychotic response to risperidone, which was based on the differential affinity of risperidone for Kv11.1 in contrast to all other drugs in the trial, we combined all other medications as one group (see the online
data supplement for more detail). Because the mean and variance of estimated drug clearance varied with different drugs, we assigned an ordinal measure of 1 to 3 according to the tertile distribution of each drug clearance to make the estimated measurements comparable between drugs. Using an ordinal measure based on the tertiles of estimated drug clearance for each drug while adjusting for the type of drugs in the same model of analysis allowed us to capture the likely nonlinear relationship between estimated drug clearance and treatment response (
9). For this analysis, however, since non-risperidone medications were all combined into one group, we considered the possibility that the effect of drug clearance using tertiles may be different between drugs and consequently may affect our assessment of the overall effect of drug clearance. Therefore, we performed a leave-one-out sensitivity analysis to assess for such a potential bias.
Results
Antipsychotic Drug Block of Kv11.1 Channel Isoforms
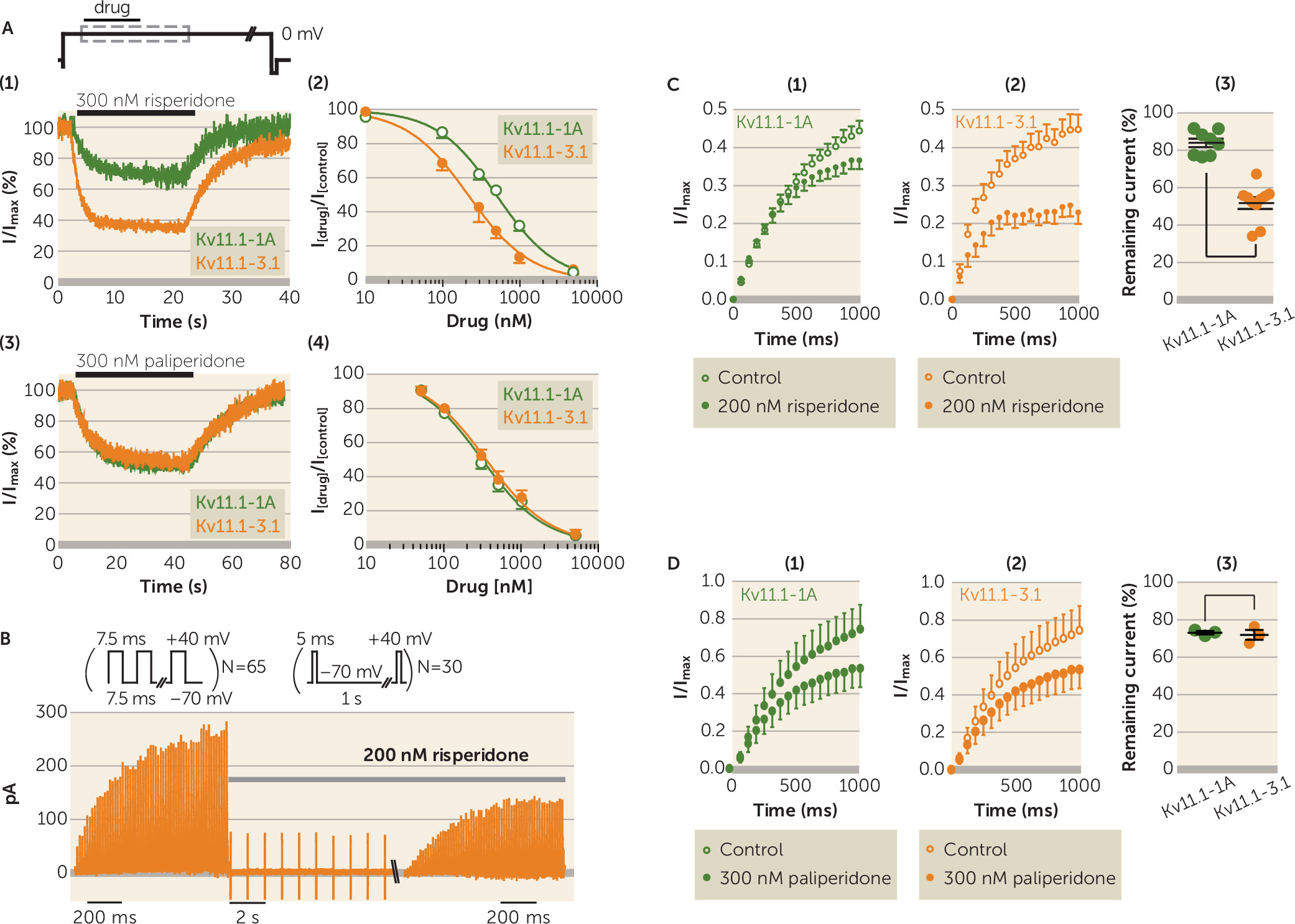
To compare the effect of antipsychotic drugs on Kv11.1 channel isoforms, Kv11.1-1A and Kv11.1-3.1 channels were expressed in Chinese hamster ovary cells and currents were recorded using whole-cell voltage clamp techniques (
13). Typical current traces recorded from Kv11.1A or Kv11.1-3.1 during the wash-on and wash-off of a range of antipsychotic drugs and the corresponding IC
50 values are reported in Figure S1 and Table S3 in the online
data supplement; the key results are presented in
Figure 1. Risperidone caused more block of Kv11.1-3.1 (IC
50=220 nM, SEM=56) compared with Kv11.1-1A channels (IC
50=508 nM, SEM=27) (
Figure 1A). In contrast, paliperidone, the main metabolite of risperidone, showed no difference in block of Kv11.1-1A and Kv11.1-3.1 channels (IC
50=307 nM [SEM=29] and IC
50=346 nM [SEM=29], respectively) (
Figure 1A).
Drug block of Kv11.1 channels may be protocol dependent (
17). We therefore investigated whether there was a similar differential block of Kv11.1-1A and Kv11.1-3.1 by risperidone compared with paliperidone or other second-generation antipsychotics when channels were stimulated with repetitive short depolarization pulses that mimic trains of neuronal action potentials.
Figure 1B shows typical current responses for Kv11.1-3.1 channels during 7.5-ms depolarization pulses to +40 mV from a holding potential of −70 mV before and 30 seconds after administration of 200 nM of risperidone. This dosage of risperidone caused 16% (SEM=2.3) block of Kv11.1-1A currents (N=8) but 48% (SEM=3.1) block of Kv11.1-3.1 currents (N=10) (p<0.001) (
Figure 1C). Conversely, 300 nM of paliperidone (N=3) caused similar block of Kv11.1-1A (26.5%, SEM=1) and Kv11.1-3.1 currents (28%, SEM=3) (p=0.70) (
Figure 1D).
KCNH2 Risk Genotypes and Response to Risperidone Treatment
Our in vitro studies demonstrate that risperidone, alone among the antipsychotic drugs tested, showed greater block of Kv11.1-3.1 than Kv11.1-1A channels. If block of Kv11.1-3.1 is a factor in antipsychotic response, as has been suggested (
9), then
KCNH2 genotype should show a stronger association with response to risperidone than to other drugs. When all 362 patients from the phase 1/1A CATIE trial for whom we have drug clearance data were considered together, irrespective of
KCNH2 SNP genotype, there was no differential treatment effect on symptoms in patients treated with risperidone compared with other drugs (see Table S4 in the online
data supplement). However, significant differences emerged when patients were classified according to
KCNH2 genotypes into three groups: diplotype group 0 (containing no minor alleles at SNPs rs3800779 and rs1036145, N=142; treated with risperidone, N=29), diplotype group 1 (containing 1–2 minor alleles at these SNPs, N=168; treated with risperidone, N=40), and diplotype group 2 (containing 3–4 minor alleles at these SNPs, N=52; treated with risperidone, N=19) (see Tables S1 and S2 in the
data supplement for detailed information on diplotype groups). The data for symptoms ratings, based on the PANSS, were analyzed using a general linear mixed model adjusting for covariates of sex, age, duration on medication, estimated drug clearance, and whether a patient completed the CATIE trial or discontinued use of medication before the end of the 18-month trial (see Table S5 in the
data supplement). Patients treated with risperidone compared with other drugs showed significant improvement in PANSS positive symptom ratings (parameter estimate=2.9, p=0.04) if they carried risk-associated diplotypes, but tended to get worse (estimate=−1.4 to −2.4) if they belonged to the other two-diplotype groups. Overall, there was a statistically significant interaction between diplotype group and drug treatment (p=0.006) (
Table 1).
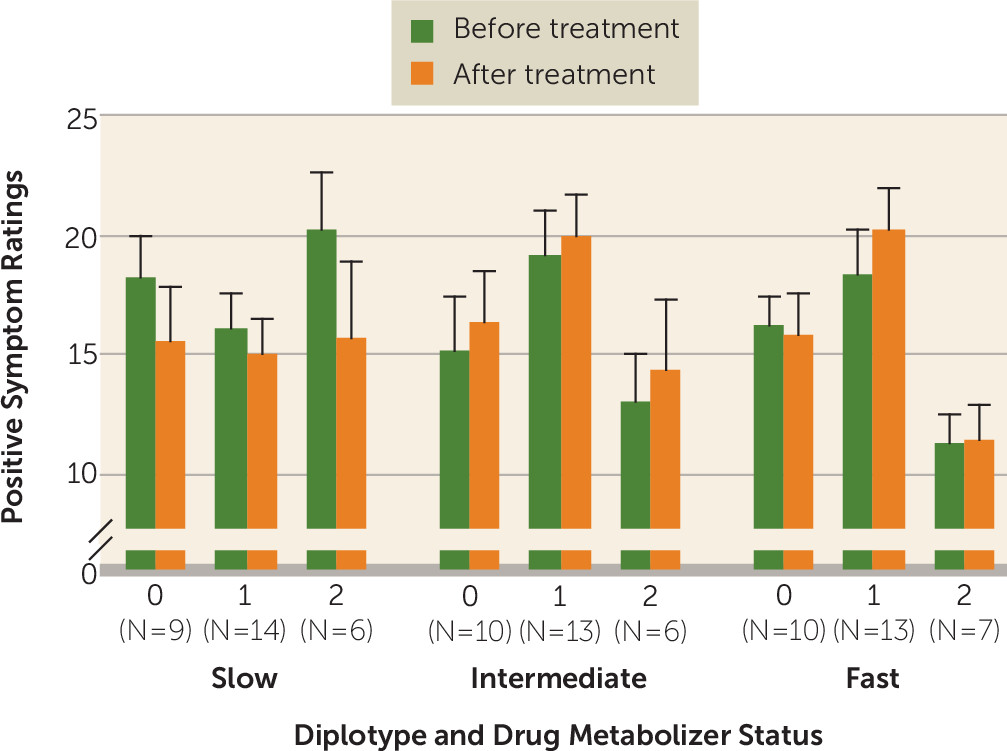
Given the recent demonstration that response to risperidone may be specifically influenced by drug metabolizer status, we further analyzed the CATIE cohort after classification into the three
KCNH2 diplotype groups as well as tertiles according to drug metabolism rate (fast, intermediate, and slow). There was no correlation between drug metabolizer status and
KCNH2 diplotype group (see Table S6 in the
data supplement) but we did find trends for three-way interactions between risperidone metabolism,
KCNH2 diplotype, and treatment (before and after the trial) on PANSS positive symptoms (p=0.07) and general psychopathology (p=0.06) (
Figure 2; see also Table S8 in the
data supplement). Specifically, patients in diplotype group 2 who had slow risperidone metabolism showed significant improvement in their PANSS positive ratings compared with the intermediate (estimate=7.0, p=0.03) and fast metabolism groups (estimate=7.25, p=0.02). We also observed that in individuals who were in diplotype group 2 and had slow metabolism, there was trend toward improvement of PANSS general psychopathology ratings compared with the intermediate metabolism group (estimate=7.43, p=0.15) and a significant improvement compared with the fast metabolism group (estimate=11.55, p=0.02) (
Figure 2; see also Table S8 in the
data supplement). Interestingly, the relationship between genotype and risperidone clearance is underscored by examining the change in ratings in the fast metabolism group. In this group, diplotype was not associated with improved response (see Table S8).
Slow drug metabolizers may be more likely to experience drug side effects and discontinue treatment because of tolerability issues (
15), although it is difficult to see how this would lead to a better response. Nevertheless, we did not find evidence of this confounder here. We found no difference in rate of discontinuation between slow metabolizers who carry the risk-associated haplotype (11/20 discontinued, 55%) compared with all other subjects (38/68 discontinued, 55.8%). Also, we were particularly interested in the risk of QT prolongation on ECG due to the association with block of Kv11.1 channels. We did not find any significant associations between risperidone clearance and change in QT interval in patients treated with risperidone in phase 1 or phase 2 of the CATIE trial (N=141 patients with ECG measurements; linear regression: r
2=0.003, p=0.99; see Figure S2 in the
data supplement).
Discussion
One of the major challenges in psychiatric therapeutics, indeed in medical therapeutics in general, is to individualize medicine to optimize response. There is currently no algorithm or clinical data to predict who will or will not respond to any particular antipsychotic drug. We now show that risperidone is unique among the antipsychotics tested as having relatively greater affinity for Kv11.1-3.1, the isoform that has increased expression in schizophrenia patients and is associated with KCNH2 genotypes, than for the cardiac-specific Kv11.1-1A isoform. Furthermore, this KCNH2 genotype specifically interacts with slow risperidone metabolism to modulate this pharmacogenetic association. It is also notable that individuals who are slow metabolizers of risperidone and have KCNH2 risk-associated diplotypes have by far the best response of anyone in the CATIE trial, whereas individuals who are not slow metabolizers and who do not have KCNH2 genotypes associated with Kv11.1-3.1 expression do not do well when treated with risperidone. While the sample sizes become small when groups are divided by diplotype and metabolism status, the data suggest that patients who have KCNH2 risk-associated genotypes and are slow metabolizers should be treated with risperidone; however, before one could make a formal recommendation to this effect, it would be necessary for the results in this study to be substantiated in a much larger efficacy study.
Previous clinical studies have demonstrated associations between treatment response to antipsychotics in general and SNPs in
KCNH2 (
9) as well as an association with rate of metabolism measured as estimated drug clearance (
9) or predicted by genotype (
11). Our study provides a mechanism for each of these associations and advances both of these earlier results to a much more biologically informed level and suggests a strategy for identifying likely risperidone responders and nonresponders. It is worth noting that in individuals with minor alleles at
KCNH2 risk SNPs who are also slow drug metabolizers, the response to risperidone treatment (an improvement of 7.0–7.2 units for PANSS ratings compared with intermediate or fast metabolizers; see Table S8 in the
data supplement) is virtually double that of the general therapeutic effect of any of the drugs tested in the CATIE trial (
7). We should caution, however, that there were only relatively small numbers of patients in the CATIE trial cohort who were taking risperidone and for whom we had drug clearance data (N=88). Almoguera and colleagues’ observation (
11) that slow metabolizer status, as determined by genetic variants in cytochrome P450 and multidrug resistance genes, may be associated with improved response to risperidone treatment does, however, provide independent replication of our clinical observations, although it should be noted that Almoguera et al. did not identify a mechanism or look at any interaction with
KCNH2 SNPs. It is also important to point out that the CATIE cohort we analyzed was part of a clinical comparative effectiveness trial (
7) rather than a pharmacogenetic trial (
18), which would be a better design to test our hypothesis.
Our in vitro analysis of drug block of Kv11.1 channel isoforms showing that risperidone, unlike its major metabolite paliperidone, preferentially blocks Kv11.1-3.1 compared with Kv11.1-1A channels provides a plausible mechanistic explanation for the clinical observations that response to risperidone treatment is influenced both by KCNH2 genotype and metabolizer status. Not only do these data have implications for pharmacogenomics and individualized therapy in schizophrenia, they also suggest that development of drugs with an even greater selectivity for inhibition of Kv11.1-3.1 channels relative to Kv11.1-1A channels could be of significant clinical benefit, particularly if they did not undergo rapid metabolism to drugs that no longer show selectivity for Kv11.1-3.1 over Kv11.1-1A channels.
Typically most antipsychotics show a 4- to 100-fold higher affinity for antidopaminergic D
2 receptors (the primary target) compared with Kv11.1-1A channels (
2,
19–
21). The therapeutic window for risperidone is 10–38 ng/mL (
22–
24), while the IC
50 for Kv11.1-3.1 and Kv11.1-1A channels (220 nM and 508 nM, respectively) corresponds to 90 ng/mL and 210 ng/mL. Based on research on plasma and brain concentrations of risperidone (
25), we would expect to see
∼5%−15% block of Kv11.1-1A channels, at the therapeutic range of risperidone concentrations, consistent with the reported QTc prolongation of 12 ms for patients treated with risperidone (
26). Conversely, given the higher efficacy of block of Kv11.1-3.1 channels, we would expect therapeutic doses of risperidone to block
∼15%−30% of Kv11.1-3.1 channels. However, based on our previous analysis of risperidone clearance levels, it is likely that in slow metabolizers, the level of risperidone will be higher than the median levels (
12). Furthermore, in the repetitive pulse protocol, risperidone was an even more efficient blocker of Kv11.1-3.1 than of Kv11.1-1A. Thus, it is possible that in this subset of patients there would be a much higher degree of Kv11.1-3.1 block during repetitive neuronal action potential firing. If block of Kv11.1-3.1 is beneficial, then there is clearly room for improvement over risperidone, which shows only a 2.5-fold greater affinity for Kv11.1-3.1 over Kv11.1-1A, and the affinity for Kv11.1-3.1 is only
∼1/10th of that for D
2 receptors. The development of more selective Kv11.1-3.1 inhibitors should also have the added benefit of reducing the potentially lethal cardiac side effects caused by inhibition of the Kv11.1-1A isoform (
27).
Conclusions
The data in this study strongly suggest that schizophrenia patients who are slow metabolizers and have KCNH2 risk-associated genotypes do better when treated with risperidone than with other antipsychotics, and they have by far the best response of anyone in the CATIE trial. Based on the numbers in this study, we estimate that ∼7% of schizophrenia patients would have the risk genotypes and slow risperidone metabolism and so would obtain the selective enhanced benefit from risperidone treatment. Conversely, the data suggest that individuals who are not slow metabolizers and do not have KCNH2 genotypes associated with Kv11.1-3.1 expression do not have a beneficial response to risperidone.
As the sample size in this study is small, it has limited power for three-way interaction analysis. Accordingly, a thorough test of our hypothesis that risk alleles in intron 2 of KCNH2 in patients who are also slow drug metabolizers will respond better to treatment with risperidone compared with other antipsychotic drugs will require a double-blind placebo-controlled crossover pharmacogenetic trial that is sufficiently powered to obtain the robust data that would be needed to inform clinical decision making. Such a trial will also require the evaluation of these results in the context of other sources of variability in the efficacy of drugs, such as nonadherence due to a drug’s neurological and endocrine side effects.
Acknowledgments
Dr. Heide is supported by a postgraduate scholarship from the Schizophrenia Research Institute; Dr. Vandenberg and Dr. Shannon Weickert are supported by Senior Research Fellowships from the National Health and Medical Research Council; and Dr. Green is supported by a Future Fellowship from the Australian Research Council. Dr. Shannon Weickert is also supported by the Schizophrenia Research Institute, the University of New South Wales, and Neuroscience Research Australia.