Cigarette smoking during pregnancy is one of the most common adverse exposures during the fetal period: approximately 12%−25% of pregnant women in Western societies smoke while pregnant (
1). Nicotine readily crosses the placenta into the fetal bloodstream, with higher concentrations than in the pregnant woman (
2). Nicotine specifically targets fetal brain development, causing short- and long-term changes in cognition (
2), neuromorphology (
3,
4), and neurotransmitter function and altered regulation of neuronal apoptosis (
5). These effects occur in part through modulation of brain nicotinic acetylcholine receptors, which have a vital role in brain maturation (
6). Moreover, prenatal nicotine exposure is related to epigenetic events (
7), dysregulation of gene transcription in placental and fetal cells (
8), and oxidative stress (
9), which adversely influence brain development. Each of these effects potentially contributes to neurodevelopmental abnormalities (
6).
Increasing evidence supports a role for pre- and postnatal environmental insults, which affect neurodevelopment in schizophrenia (
10). Studies of maternal smoking as a risk factor for schizophrenia in offspring have revealed contradictory findings, and most have had significant limitations. In a Finnish birth cohort study, limited by a small sample size, no association with schizophrenia was observed (
11). In two other studies, which examined schizophrenia as an outcome, results were conflicting (
12,
13). In those studies, data on maternal smoking during pregnancy were acquired retrospectively, by maternal interview, which can be limited by recall bias. Moreover, clinical, rather than population-based, samples were used (
12,
13). A Finnish register-based study found an association between maternal self-reported smoking and psychotic disorders in general (
14). The remaining studies, which reported associations between maternal smoking and psychotic symptoms in adolescents, did not examine schizophrenia per se (
15,
16). We know of no previous study to date on maternal smoking and schizophrenia that used biomarker data to define the exposure.
We examined whether maternal smoking was related to schizophrenia in the Finnish Prenatal Study of Schizophrenia. Unlike all previous studies, our investigation utilized maternal cotinine levels measured in prospectively drawn, biobanked maternal serum specimens during early to mid-gestation. Moreover, we employed a national registry-based study with nearly 1,000 cases of schizophrenia, which addressed the limitations of small sample sizes and use of clinical samples in prior studies. We hypothesized that increasing maternal cotinine levels during pregnancy are associated with an increased odds of schizophrenia in offspring.
Method
The Finnish Prenatal Study of Schizophrenia is based on a nested case-control design. The sampling frame was defined such that all birth cohort members were within the age of risk for schizophrenia. Hence, the sample consisted of all offspring born in Finland from 1983 to 1998, and the subjects were followed up until 2009 (see “Case and Control Identification” below).
Description of Cohort and Biobank
All offspring in the Finnish Prenatal Study of Schizophrenia were derived from the Finnish Maternity Cohort, which consists of virtually all pregnancies in Finland with archived prenatal serum specimens drawn beginning in 1983 (total number of samples is over 1 million). Sera were drawn during the first and early second trimesters (5th to 95th percentile: months 2–4 of pregnancy) from over 98% of pregnant women in Finland, following informed consent, for screening for HIV, syphilis, and hepatitis. One maternal serum sample was obtained for each pregnancy. Serum samples were stored as one aliquot at −25°C in a single, centralized biorepository at the National Institutes of Health and Welfare. All serum samples in the Finnish Maternity Cohort can be linked with offspring by a unique personal identification number (PIN), which has been assigned to each resident of Finland since 1971.
Finnish Population Registry
The computerized nationwide registry includes comprehensive data on place of birth, date of emigration, date of death, place of residence, and biological parents, including their birth dates.
Case and Control Identification
The Finnish Hospital and Outpatient Discharge Registry was used to identify all recorded diagnoses for psychiatric hospital admissions and outpatient treatment visits among cohort members. The registry contains the personal and hospital identification codes and psychiatric diagnoses. Computerized data are available from 1987 to the present.
To identify the cases, we conducted a record linkage between the Finnish Maternity Cohort and the discharge registry, using the PINs. Cases with schizophrenia (ICD-10 F20) or schizoaffective disorder (ICD-10 F25) were followed up until 2009 (these cases are referred to as “schizophrenia”). The age at first treatment was recorded by the first contact with a psychiatric facility with a schizophrenia diagnosis. The validity of schizophrenia diagnoses according to the discharge registry was excellent; 93% of subjects with a diagnosis of schizophrenia in the discharge registry were assigned a consensus diagnosis of schizophrenia (
17). There were 977 cases with sufficient serum volume for the cotinine assays; these were included in all analyses.
The case subjects were matched 1:1 to control subjects drawn from the birth cohort who were without schizophrenia, other nonaffective psychotic disorders, and bipolar disorder. They were matched on date of birth (within 1 month), sex, and residence in Finland at the time of case diagnosis. A flow chart of subject selection is shown in Figure S1 in the data supplement accompanying the online version of this article.
The study was approved by the ethical committees of the Hospital District of Southwest Finland, the National Institutes of Health and Welfare, and the Institutional Review Board of the New York State Psychiatric Institute. Informed consent was obtained prior to acquisition of maternal serum specimens.
Cotinine Assay
Cotinine measurements were conducted by researchers blind to case/control status. Serum cotinine levels were measured using a commercially available quantitative immunoassay kit (OraSure Technologies, Bethlehem, Pa.) (sensitivity=96%−97%, specificity=99%−100%). Intra- and interassay variation are 3.5%−6.2% and 6.0%−9.6%, respectively. The limit of detection was 0.08 ng/ml.
Covariates
The covariates included sex, maternal age, paternal age, socioeconomic status (maternal education), province of birth, municipality of birth (urban, semiurban, rural), parental history of schizophrenia and other psychiatric disorders, previous births, weight for gestational age, maternal C-reactive protein (CRP) level, and gestational week of the maternal blood draw. All covariates except gestational age and gestational week of the blood draw were obtained from the Finnish Population Registry. Gestational age was obtained from the Finnish Medical Birth Register and obstetric record review, gestational week of the blood draw was obtained from the Finnish Maternity Cohort, and maternal CRP level was obtained from assays of the archived maternal serum specimens. In accord with the extant epidemiologic literature, covariates were included in the statistical models based on associations with both maternal cotinine exposure (p≤0.1) and schizophrenia (p≤0.1).
Statistical Analysis
The analysis was based on a nested case-control design, in which the control subject for each case was drawn from the population at risk (the Finnish Prenatal Study of Schizophrenia birth cohort) and matched on selected factors (see “Case and Control Identification”). In the primary analysis, we examined cotinine as a continuous measure. Given the skewed distribution of cotinine levels, the variable was log-transformed before analysis. To account for missing cotinine levels below the limit of detection (0.08 ng/ml), we constructed a conditional logistic regression model with the following predictors: 1) an indicator of a cotinine level below the level of detection and 2) the indicator of the complementary event multiplied by log(c+cotinine level), where c is a small positive constant. In the results of the analyses presented below, c was set to 0. Sensitivity analyses were used to assess how the results changed as c varied between the limit of detection (LOD) and 3*LOD; the choice of c within this range had little effect on the results (results presented on request).
To further facilitate interpretation of the data, we then examined maternal cotinine as a three-class categorical variable: reference (<20 ng/ml), moderate exposure (20–50 ng/ml), and heavy exposure (>50 ng/ml). These cut-off points were recommended by the cotinine immunoassay kit and have also been used in previous studies based on the Finnish Maternity Cohort serum bank (
18). These levels also corresponded well with the frequencies of self-reported maternal smoking in mothers of case subjects (data on self-reported smoking available for 656 mothers). Smoking was reported by 22 of 508 (4.3%) mothers in the reference category, 11 of 19 (57.9%) mothers with cotinine levels of 20–50 ng/ml, and 105 of 129 (81.4%) mothers with cotinine levels >50 ng/ml (p<0.001). Appropriate to the nested case-control study design, point and interval estimates of odds ratios were obtained by fitting conditional logistic regression models for matched pairs.
We tested for interaction between maternal cotinine and a three-level variable for parental history of psychiatric disorders (schizophrenia or other nonaffective psychosis, any psychopathology, no psychiatric disorders) on the multiplicative scale using the statistical modeling approach described in the previous paragraph. We also tested for interaction between these variables on an additive scale by computing the relative excess risk due to interaction (
19). Statistical significance was based on p<0.05. Statistical analyses were performed with SAS software (SAS 9.4, SAS Institute, Cary, N.C.).
Results
The mean age of the case and control subjects was 22.3 (SD=2.2, range=14.6–25.6). Maternal and paternal age, parental history of psychiatric disorders, and province and municipality of birth were associated with schizophrenia in offspring (
Table 1). Low weight for gestational age was not associated with schizophrenia. Maternal cotinine level was associated with maternal age, maternal education, parental history of psychiatric disorders, previous births, gestational week of the blood draw, and province of birth. Maternal cotinine level was not related to gestational age (
Table 2). Maternal age, parental history of psychiatric disorders, and province of birth were associated with both schizophrenia and maternal cotinine level, and they were included in the adjusted models (
Table 3). In additional models, weight in relation to gestational age was also adjusted to test for a potential mediating effect of this covariate (see below).
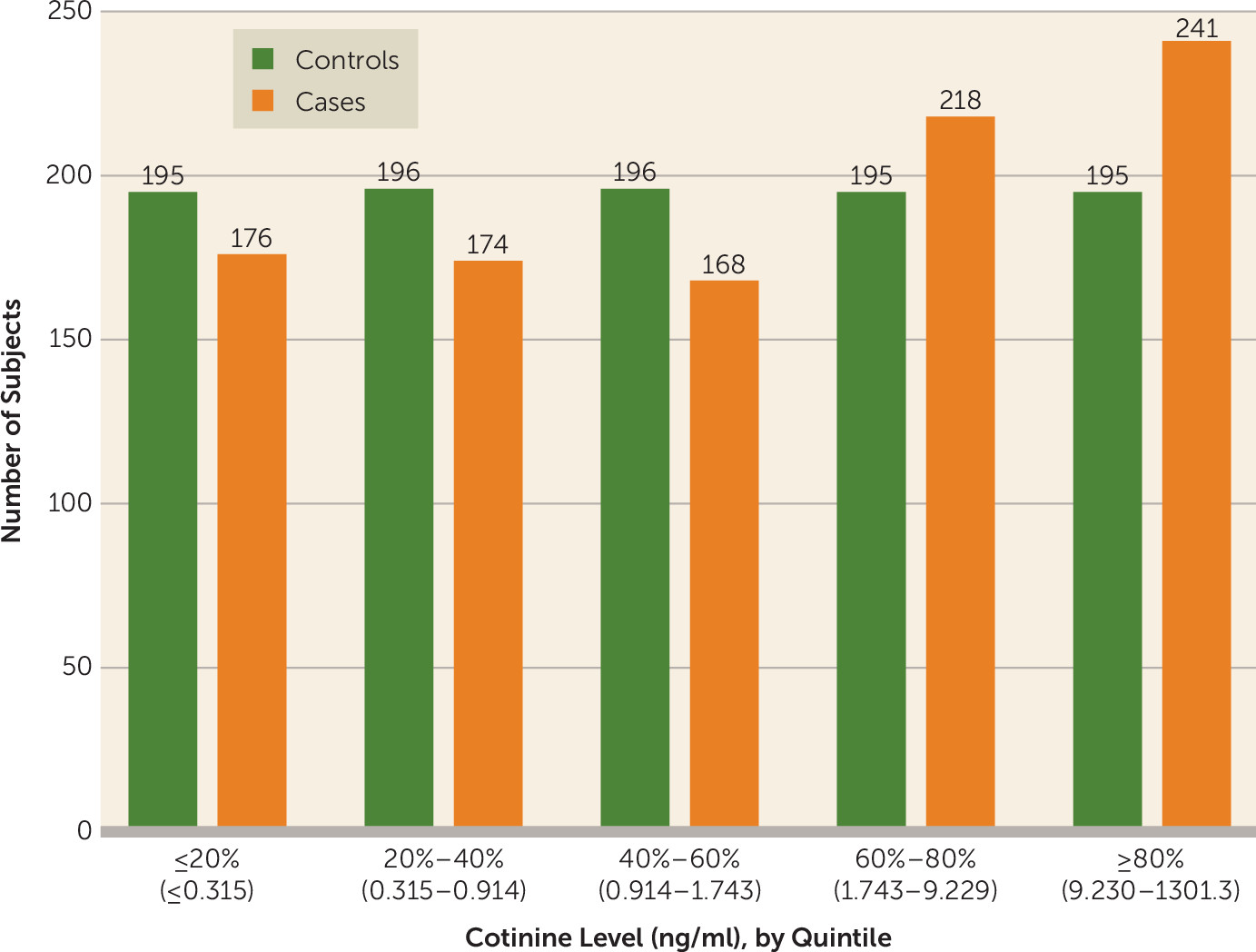
The distribution of cotinine by case-control status and exposure levels in quintiles is presented in
Figure 1. The mean cotinine level in the case subjects was 35.9 ng/ml (SD=92.9), with a range of 0.01–1301.3 and interquartile range of 0.5–8.1, and in the control subjects it was 23.1 ng/ml (SD=61.1), with a range 0.007–537.3 and interquartile range 0.4–2.8).
Main Results
The findings for the conditional logistic regression analyses are presented in
Table 3. In the unadjusted analysis, with cotinine examined as a continuous variable and taking the presence of cotinine levels below the limit of detection into account, there was a significant association between increasing log-transformed maternal cotinine and odds of schizophrenia (odds ratio=2.69, 95% CI=1.54–4.69, p<0.0001). In models 1–3 adjusting for covariates associated with maternal cotinine exposure (p<0.1) and schizophrenia (p<0.1), the odds for offspring schizophrenia associated with log-transformed maternal cotinine remained significant. In the model including maternal age, province of birth, and any parental psychiatric disorder as covariates the odds ratio was 3.41 (95% CI=1.86–6.24, p≤0.0001) (
Table 3).
In the categorical analyses (
Table 3), in which cotinine was categorized as a three-class variable, heavier nicotine exposure (cotinine level >50 ng/ml) was associated with offspring schizophrenia after adjustment for covariates (models 1–3), including any parental psychiatric disorder (odds ratio=1.38, 95% CI=1.05–1.82, p=0.02). Moderate nicotine exposure (20–50 ng/ml) was associated with offspring schizophrenia after adjusting for maternal age, maternal schizophrenia, and province (model 1: odds ratio=1.96, 95% CI=1.04–3.70, p=0.04) but not in the other multivariate models. However, the numbers of case and control subjects were very small in this category (29 case subjects, 20 control subjects).
Supplemental Results
For further reassurance, we conducted supplemental analyses. First, we excluded schizoaffective disorder to examine whether maternal cotinine was associated with schizophrenia only. Heavier nicotine exposure remained significantly associated with schizophrenia (odds ratio=1.44, 95% CI=1.09–1.89, p=0.009). The association persisted in the analysis adjusting for maternal age, parental schizophrenia, and province of birth (odds ratio=1.40, 95% CI=1.04–1.89, p=0.03). Maternal cotinine examined as a continuous variable was also associated with schizophrenia only, adjusting for these covariates (odds ratio=2.91, 95% CI=1.48–5.70, p=0.02).
Second, given that smoking is a known cause of low weight relative to gestational age and that high cotinine was related to low weight in this study, the latter variable may be considered as a potential mediator of the relationship between maternal smoking and schizophrenia. To test for mediation, we added weight for gestational age to the models. In each of the models in which weight was adjusted, the odds ratios for low weight and schizophrenia were all below 1 (
Table 4). Given that a key criterion for establishing a covariate as a mediator in a putative causal pathway is a relationship with the outcome (
20) in the model adjusting for this covariate, this lack of association indicates that low weight for gestational age did not mediate the association between maternal cotinine and schizophrenia. Moreover, in the models in which cotinine was classified as a continuous variable, there were generally modest changes in the odds ratios from those presented in
Table 3; for cotinine classified as a categorical variable, the addition of weight led to a modest decrease in the odds ratios, ranging from 9.2% to 17.3% (
Table 4). There was one notable change (44% decline) in the odds ratio following adjustment for weight (model 3). However, there was a statistical trend in the p value (p=0.06) in the adjusted model, indicating a persistent association between maternal cotinine and schizophrenia.
Maternal self-reported smoking was also associated with schizophrenia. Among subjects for whom maternal smoking data were available, 138 of 656 (21.0%) mothers of case subjects smoked, while among the control subjects, 106 of 670 (15.8%) mothers of control subjects smoked (p<0.01).
Test for Interaction Between Maternal Cotinine and Parental Psychiatric Disorders
We tested for interaction between a three-level variable for parental history of psychiatric disorders and maternal cotinine on the multiplicative scale by adding four product terms for the interaction of parental history and cotinine to the adjusted conditional logistic regression model. The odds ratios for schizophrenia associated with heavy and moderate versus low cotinine exposure were 1.22 (95% CI=0.46–3.26, p=0.69) and 0.95 (95% CI=0.08–11.35, p=0.97) among those with a parental history of schizophrenia or nonaffective psychosis, 1.82 (95% CI=1.11–2.98, p=0.02) and 2.94 (95% CI=0.75–11.56, p=0.12) among those with a parental history of any psychiatric disorders, and 1.30 (95% CI=0.91–1.85, p=0.16) and 1.73 (95% CI=0.77–3.89, p=0.18) among those with no parental history of psychiatric disorders. However, there was no statistical evidence for interaction on the multiplicative scale. None of the product terms for the interaction of parental history and cotinine exposure was statistically or marginally significant (p>0.2), and the group of four terms did not contribute significantly to the model (p=0.75).
We computed the relative excess risk due to interaction (RERI) to test for the effect of additive interaction between parental history of psychiatric disorders and categorically defined maternal cotinine levels on the risk of schizophrenia. Considering those with a parental history of schizophrenia or nonaffective psychosis versus no family history of psychiatric disorders, the RERI for heavy versus low maternal cotinine exposure was above 0 but not statistically significant (RERI=1.92, 95% CI=−7.31 to 11.14, p=0.68). For moderate versus low maternal cotinine, the RERI was less than 0 and fell short of statistical significance (RERI=−5.69, 95% CI=−11.80 to 0.43, p=0.07). Comparing parental history of other psychiatric disorders to no family history, we found that for both heavy levels (RERI=1.08, 95% CI=−0.34 to 2.50, p=0.14) and moderate levels (RERI=2.91, 95% CI=−4.57 to 10.39, p=0.45) versus low levels of maternal cotinine, RERI estimates were above 0 but not statistically significant.
Discussion
In this nationwide population-based nested case-control study of nearly 1,000 schizophrenia subjects, we demonstrated an association between maternal nicotine exposure, quantified as cotinine during gestation, and schizophrenia in offspring. For cotinine classified as a continuous variable and for heavy nicotine use, the association persisted after adjusting for covariates. There was no clear evidence of mediation of this relationship by low weight for gestational age, particularly since it was not a risk factor for schizophrenia in this population. Moreover, we did not find convincing evidence for interaction between parental schizophrenia or other psychiatric disorders and maternal cotinine on a multiplicative or additive scale. To our knowledge, this is the first biomarker-based study to show a relationship between fetal nicotine exposure and schizophrenia.
The plausibility of these findings is supported by an extensive literature on prenatal smoking and neurocognition. Offspring of mothers who smoke have delayed psychomotor and mental developmental scores; deficits in sustained attention, verbal learning, and design memory; impaired speech and language; and lower IQ (
2). Low premorbid IQ and other neurocognitive abilities have been related to schizophrenia (
21).
There are several potential mechanisms by which nicotine alters fetal brain development (
3,
6) so as to increase risk of schizophrenia. Nicotine binds to neuronal nicotinic acetylcholine receptors, which are essential for proper brain organization during the prenatal period (
6). Prenatal nicotine treatment decreases the number of cells in whole brain and particular subregions in the fetal and neonatal periods (
22), consistent with apoptotic processes. The effects of nicotine-induced dysregulation of neurotransmitter systems and neuromorphology persist beyond the fetal period (
6). In humans, prenatal smoking has been associated with structural brain changes in adolescence, including cortical thinning (
23) and decreased corpus callosal volume (
4). These brain anomalies have also been demonstrated in schizophrenia (
24), as have abnormalities of prenatal brain development (
10) and dopaminergic function (
25).
Prenatal nicotine is associated with abnormal development of cerebral inhibitory neurons, a key pathophysiological defect in schizophrenia (
26). Neonates of mothers who smoke during pregnancy have abnormal inhibitory gating of the P1 auditory evoked response, indicating impaired cerebral inhibition. The principal cholinergic receptor involved is the α
7-nicotinic acetylcholine receptor (nAChR) (
27). A single nucleotide polymorphism in CHRNA7, the α
7-nAChR gene, is associated with schizophrenia (
28), and the CHRNA7 genotype has been linked to familial transmission of the P1 sensory gating deficiency (
29).
Nicotine also causes uteroplacental underperfusion and increases carboxyhemoglobin levels, with higher fetal than maternal levels, both of which reduce oxygen availability and its delivery to fetal tissues, resulting in hypoxia (
2). Although prenatal nicotine exposure is associated with disrupted intrauterine growth (
30,
31) and lower weight for gestational age (
32), in this study there was not strong evidence for a mediating role of low weight for gestational age.
Moreover, maternal cigarette smoking has been associated with epigenetic effects indicating sensitivity of the methylome (
33) and down-regulation of brain-derived neurotrophic factor (BDNF) (
34), which is related to schizophrenia (
35). Maternal smoking is also associated with abnormal DNA methylation patterns, which may adversely affect fetal development (
8,
33). Tobacco may also cause oxidative stress in the prefrontal cortex, hippocampus, and striatum in rodents (
9).
The study has several strengths, particularly compared with previous studies, reviewed above. These include use of the biomarker cotinine, a prospectively documented and reliable metabolite of nicotine, which obviates self-report bias and retrospective recall. The study focused on schizophrenia and schizoaffective disorder, rather than broader and less stringent definitions, including self-reported psychotic symptoms. We also employed a nationwide sample with the highest number of schizophrenia cases to date in a study of this type.
Limitations of the study include the following. First, there is comorbidity between smoking and alcohol use (
1). Unfortunately, data on maternal alcohol use during pregnancy were not available. However, the maternal psychiatric history variable, adjusted for in the multivariate analyses, included alcohol and other substance use disorders from the hospital discharge register. Second, nicotine is one of many potentially toxic compounds in tobacco (
36) that could alter early neuronal development, but biomarker data on other toxic compounds were not available. However, as noted above, there is considerable evidence that nicotine is a biologically plausible disruptor of fetal development (
36). Third, although there was no evidence of confounding following testing of numerous covariates, residual confounding may have occurred. Of note, we were able to adjust for schizophrenia in parents but not in other classes of relatives, which would have provided better control of genetic influences. Parenthetically, causal interpretations are not warranted given that other unmeasured maternal factors, including diet and life events, may have contributed to the relationship. However, these factors would need to be related to maternal smoking and schizophrenia to be confounders. Fourth, the sample was relatively young (median age 23.5), and thus further follow-up into later ages at onset will be necessary to determine whether the relationship is generalizable to these cases. Fifth, we cannot disentangle prenatal from postnatal nicotine exposure due to secondhand smoke, since no data are available on postnatal smoking in mothers or fathers. However, nicotine levels from secondhand smoke would be considerably lower than levels from fetal exposure to nicotine. Sixth, we do not have information on offspring nicotine use, which is associated with prenatal nicotine exposure (
37) and may also increase the risk of offspring schizophrenia (
38). Seventh, self-reported smoking was recorded as a binary variable only, and no data were available to correlate cotinine level and the number of cigarettes smoked. However, in a prior study using archived prenatal maternal serum samples from the Finnish Maternity Cohort (
39), a correlation was found between the cotinine level in serum and the self-reported number of cigarettes smoked daily. Finally, we did not observe a dose-response effect in the relationship between maternal cotinine in the categorical analysis and schizophrenia. However, the sample size in the group with moderate cotinine exposure was very low compared with the high-exposure group, which may have contributed to a more imprecise odds ratio (wide confidence intervals).
Conclusions
Prenatal nicotine exposure during gestation was related to an increased odds of schizophrenia. Given the high frequency of smoking during pregnancy, these results, if replicated, may ultimately have important public health implications for decreasing the incidence of schizophrenia. Further studies are necessary to address potential residual confounders. More work is also needed on maternal smoking and other environmental, genetic, and epigenetic factors. Finally, it will be of interest in future studies to examine maternal cotinine in relation to bipolar disorder, autism, and other psychiatric disorders.