Cognitive dysfunction is a core and clinically critical feature of schizophrenia (
1) but responds poorly to available medications (
2). Therefore, identifying the neural substrate for these cognitive deficits is critical for the development of new therapeutic interventions. Certain cognitive deficits, such as impaired working memory, appear to emerge from altered gamma oscillations in the dorsolateral prefrontal cortex (DLPFC) (
3). Because cortical gamma oscillations require the activity of parvalbumin-containing (PV) interneurons (
4,
5), deficient cortical PV interneuron activity could provide the neural substrate for altered prefrontal gamma oscillations and consequently impaired working memory in schizophrenia.
Lower glutamatergic drive to PV interneurons has been hypothesized to be the cause of deficient PV interneuron activity in schizophrenia (
6–
8). This hypothesis is based on findings that experimental manipulations in model systems that reduce glutamatergic drive to PV interneurons result in lower PV interneuron activity accompanied by lower expression of activity-dependent gene products (e.g., PV and the GABA-synthesizing enzyme glutamic acid decarboxylase 67 [GAD67]), abnormal gamma oscillations, and working memory deficits (
9–
12). Consistent with this hypothesis, postmortem studies have repeatedly shown lower PV and GAD67 levels in the DLPFC of schizophrenia subjects (
13–
17), which are thought to reflect lower glutamatergic drive to a subset of PV neurons and not a loss of PV neurons in the illness (
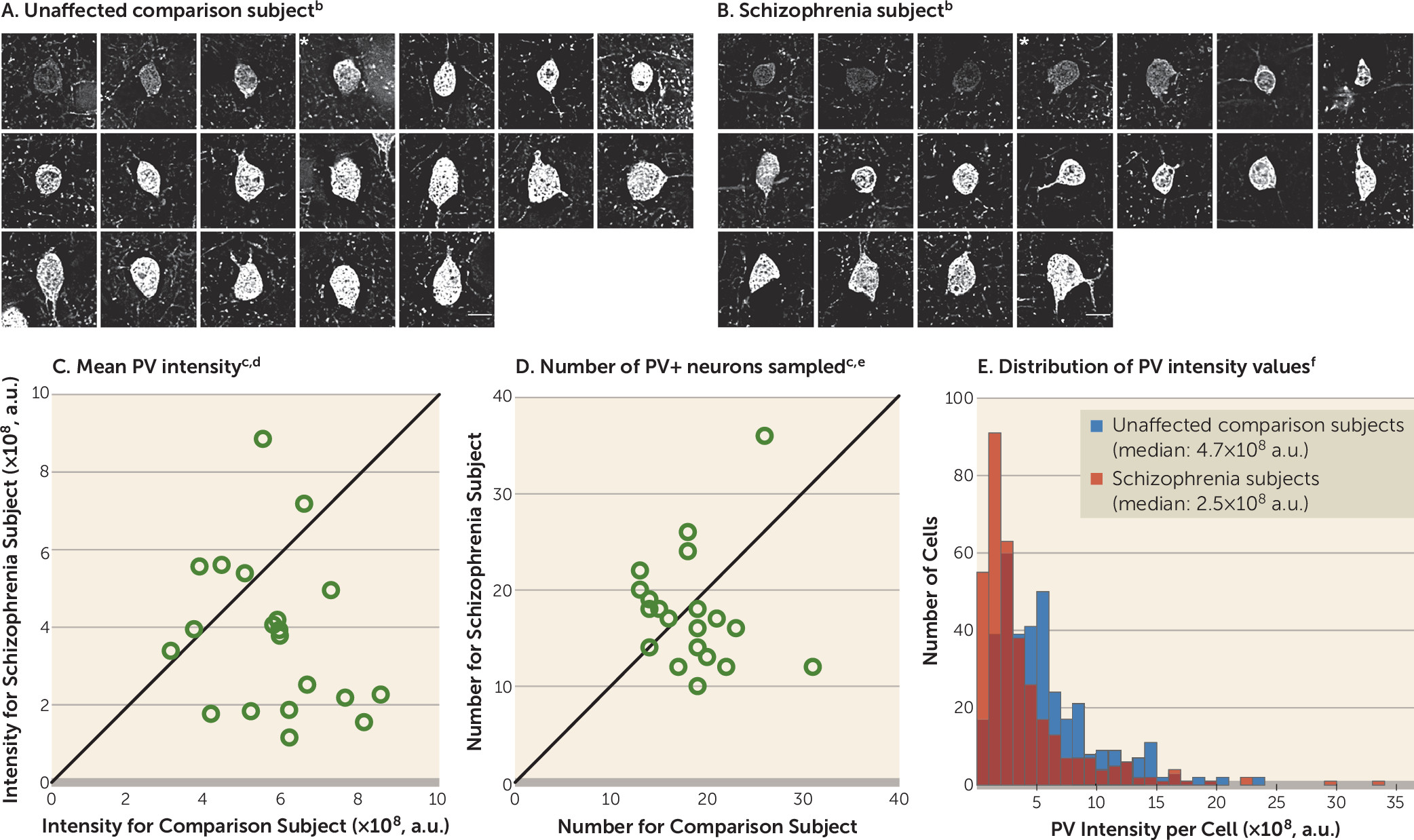
13,
15,
18,
19). These deficits do not seem to be due to a global reduction in excitatory drive to all interneuron subtypes, as calretinin-containing (CR) interneurons, the most abundant interneuron subtype in the primate DLPFC (
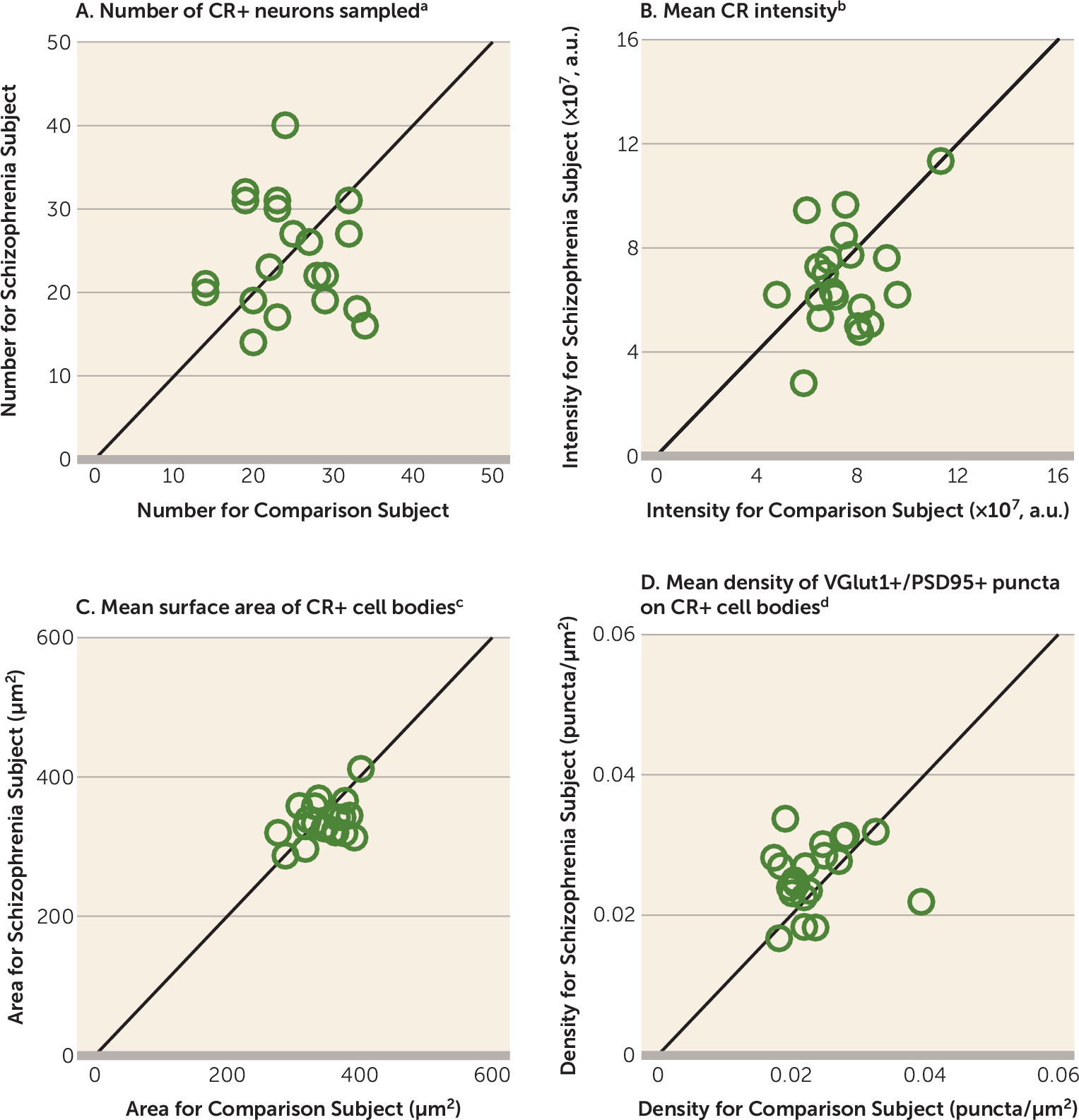
20), appear to be relatively unaffected in schizophrenia (
15,
16,
21). However, the pathological basis for lower glutamatergic drive selectively onto PV interneurons, such as fewer excitatory synapses on these neurons, has not been identified in people with schizophrenia.
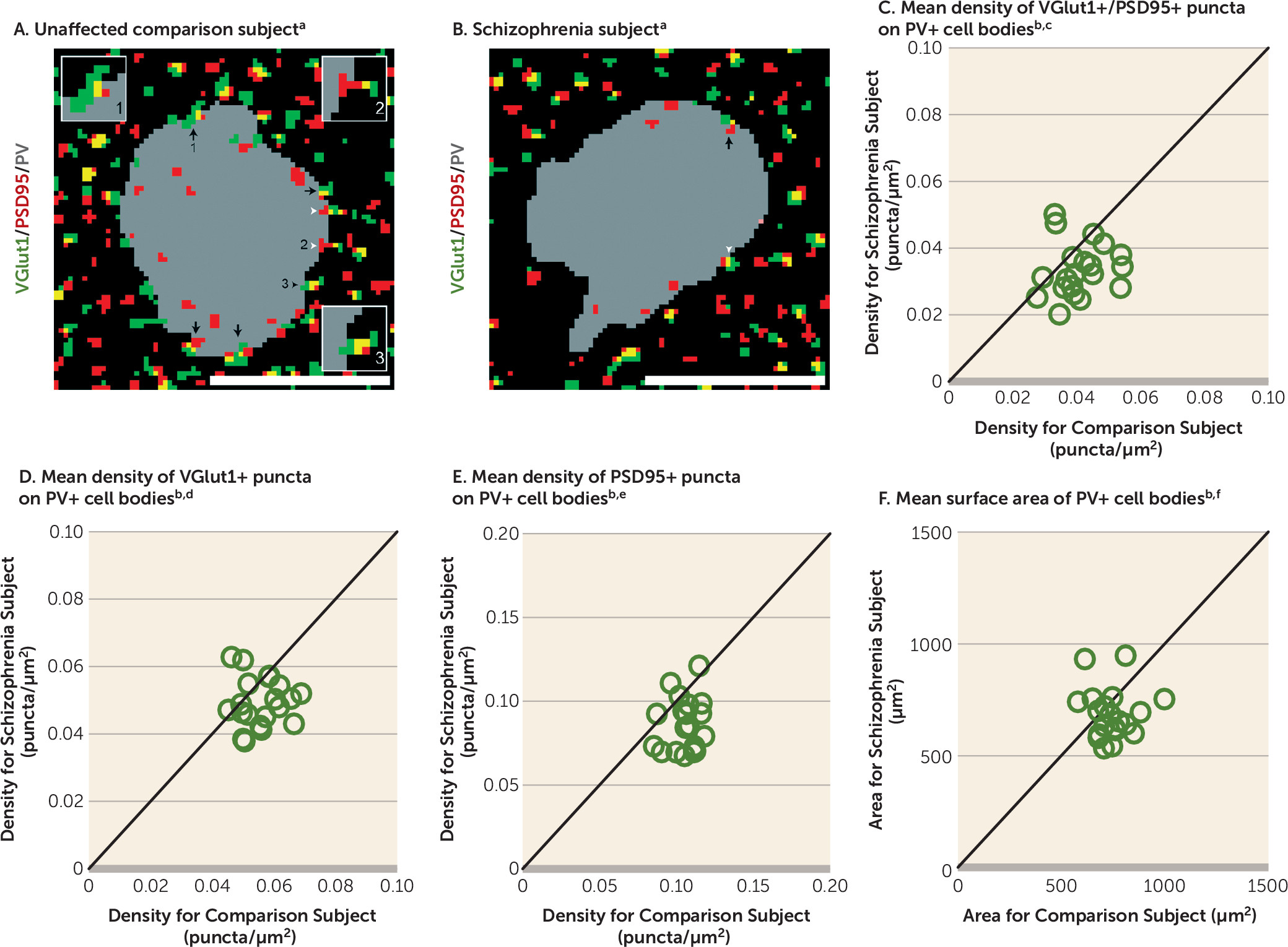
Therefore, in this study, we used multilabeling fluorescent immunohistochemistry, confocal imaging, and a custom post-image processing method to directly assess excitatory synapses on parvalbumin-positive (PV+) neurons in the DLPFC from matched pairs of schizophrenia and unaffected comparison subjects. We tested the hypotheses that 1) lower PV levels in subjects with schizophrenia reflect altered PV expression per neuron and not a loss of PV+ neurons, 2) PV+, but not calretinin-positive (CR+), neurons receive fewer excitatory synaptic inputs in subjects with schizophrenia, and 3) fewer excitatory synapses on PV+ neurons are associated with activity-dependent down-regulation of PV and GAD67 levels.
Discussion
A pathological substrate for reduced glutamatergic drive onto PV interneurons in schizophrenia has not been previously identified due to the technical challenges of resolving synaptic abnormalities in a cell type–specific manner in postmortem human brain. Here we report for the first time, to our knowledge, that the neuropathology of schizophrenia includes a lower number of excitatory synapses on PV interneurons. This deficit appears to reflect the disease process of schizophrenia and is not due to methodological confounds or other factors commonly associated with the illness. First, the presence of fewer excitatory synapses on PV+ neurons was evident from lower numbers of both pre- and postsynaptic markers, validating deficits in the synaptic structures. Second, these deficits were not confounded by either a larger surface area of PV+ cell bodies or undetectable levels of protein markers within existing synaptic structures in schizophrenia subjects. Third, none of the assessed schizophrenia-associated comorbid factors accounted for these deficits. Fourth, long-term exposure to antipsychotic medications did not alter the density of excitatory synapses on PV+ neurons in the DLPFC of nonhuman primates; the effect of antipsychotics could not be assessed in humans, as only one schizophrenia subject had not been exposed to these medications. Fifth, the density of excitatory synapses on CR+ neurons was not lower in the illness, consistent with a prominent pathology of PV interneurons in schizophrenia. Finally, the density of excitatory synapses on PV+ neurons predicted levels of the activity-dependent gene products PV and GAD67 selectively in the schizophrenia subjects. Together, these findings support the hypothesis that fewer excitatory synapses selectively on cortical PV interneurons provide a pathological substrate for deficient excitatory drive to these interneurons in schizophrenia.
PV interneurons comprise two main subtypes: PV basket neurons target the proximal dendrites and cell bodies of pyramidal neurons, and PV chandelier neurons synapse onto the axon initial segment of pyramidal neurons (
36). Although both populations of PV interneuron subtypes are active during gamma oscillations, gamma rhythms are more strongly coupled to the activity of PV basket neurons (
37,
38). Given the much greater prevalence of PV basket neurons in the middle layers of the primate DLPFC (
39,
40), most of the PV+ neurons sampled in our study are likely to be PV basket neurons.
Our sampling of excitatory synapses on PV+ neurons is limited in two respects. First, we did not sample VGlut2-containing excitatory inputs that represent projections from the thalamus (
41). However, thalamic axons represent a small minority (<10%) of excitatory terminals in the cortex (
42), and only a small percentage of thalamic inputs target PV interneurons (
43), suggesting that the excitation of PV interneurons is mostly driven by cortical excitatory inputs. Second, we did not sample synaptic inputs to dendrites of PV interneurons due to the few dendrites with detectable PV immunoreactivity that originate from PV+ cell bodies. Although the density of excitatory synapses is higher on dendrites than cell bodies of PV interneurons (
44), depolarization of the cell body of PV interneurons is much stronger for somal than dendritic excitatory inputs (
45,
46). However, despite these limitations, the density of excitatory cortical synapses on PV+ cell bodies in the present study predicted activity-dependent PV expression across all PV+ neurons. Therefore, our approach was sufficiently sensitive to detect an apparently functionally meaningful deficit in excitatory synaptic inputs to PV+ neurons in schizophrenia subjects.
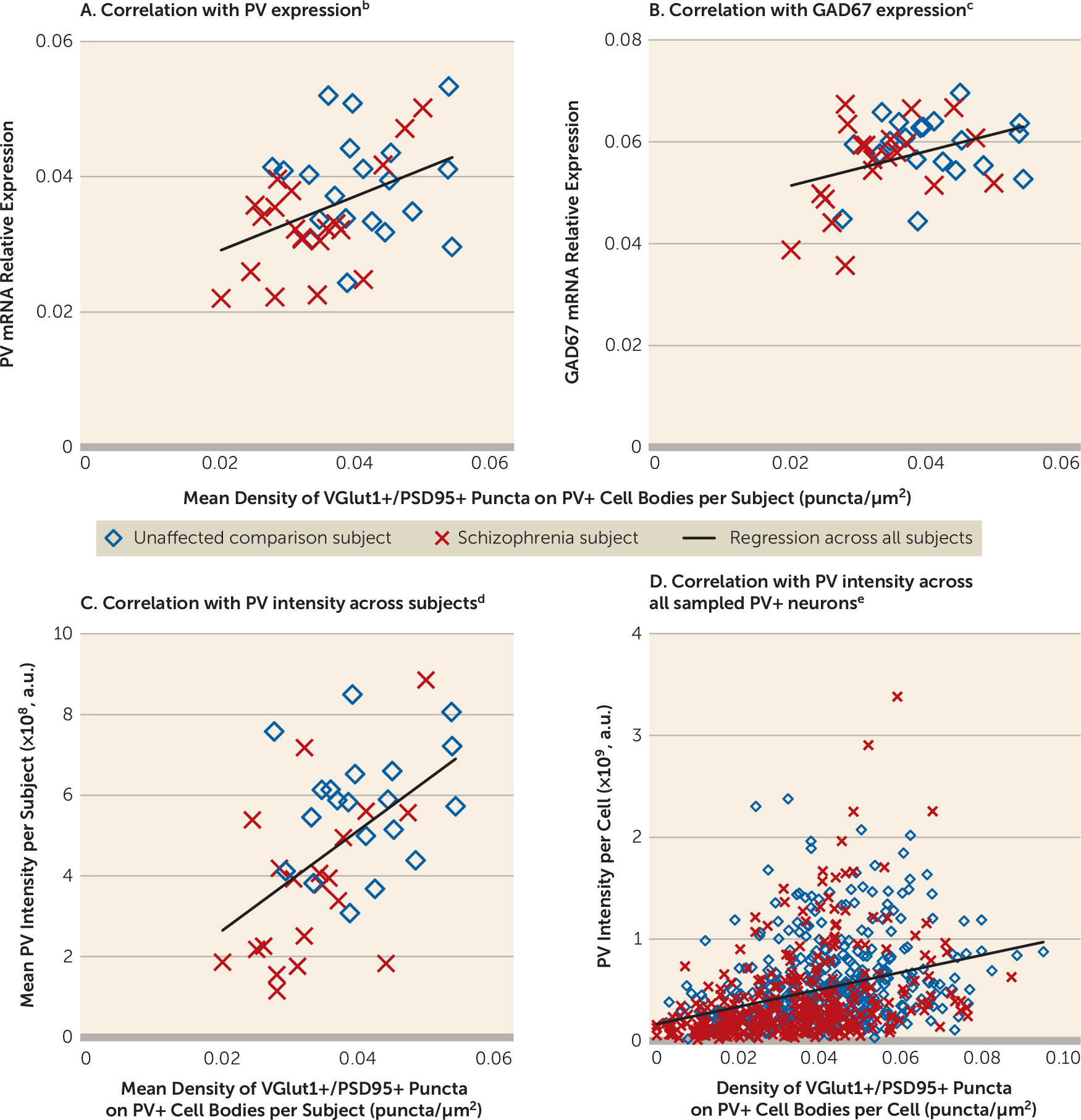
Multiple signaling pathways regulate the formation of excitatory synapses on PV interneurons. For example, the ErbB4 signaling pathway induces the formation of excitatory synapses on PV interneurons (
11,
47,
48), and the release of neuronal pentraxin 2 (Narp) from pyramidal cells recruits excitatory synapses selectively on PV interneurons in an activity-dependent manner (
28). Both an abnormal shift in ErbB4 splicing at the JM locus (
49,
50) and lower Narp transcript levels have been shown in the DLPFC of subjects with schizophrenia, including those included in the present study (
21,
51). Across these subjects, the density of VGlut1+/PSD95+ puncta on PV+ cell bodies was significantly correlated with the ratio of ErbB4 JM-a to JM-b splice variants in cortical layer 4 (r=−0.44, p=0.005) and with Narp mRNA levels in total gray matter (r=0.35, p=0.03). These findings suggest that alterations in the ErbB4 and/or Narp signaling pathways could be upstream of the deficit in excitatory synaptic inputs to, and consequently the lower activity of, PV interneurons in schizophrenia.
Consistent with the idea that PV interneuron activity is reduced in the illness, activity-dependent expression levels of GAD67 are lower in the DLPFC of subjects with schizophrenia, and specifically in PV interneurons (
15,
17). Experimental reductions of GAD67 in PV interneurons decrease inhibitory synaptic transmission in pyramidal cells and alter cortical network activity (
52–
54). Strong inhibition of pyramidal cells by PV interneurons is required for the generation of gamma oscillations in the DLPFC associated with working memory. Thus, the lower density of excitatory synapses on PV+ neurons and the corresponding deficit in GAD67 expression found in the present study could provide a pathological substrate for deficient inhibition of pyramidal cells by PV interneurons, which in turn would result in impaired gamma oscillations and working memory deficits in people with schizophrenia.
Discovering pathological entities that bridge etiopathogenic pathways to the core features of the illness is essential for understanding the disease process of schizophrenia. For example, disruptions in the ErbB4 or Narp signaling pathway in the DLFPC could be upstream of the deficits in excitatory synaptic inputs to PV interneurons in schizophrenia. Given that PV interneuron activity is essential for gamma oscillations, these deficits could underlie the downstream pathophysiology of impaired gamma oscillations and consequently working memory dysfunction in schizophrenia. Therefore, fewer excitatory synapses on PV interneurons might serve as a common pathological locus upon which diverse streams of etiopathogenic pathways converge in order to produce a core pathophysiological feature of schizophrenia from which cognitive dysfunction emerges.