Affective disorders, with depression leading the way, are a major contributor to the morbidity caused by diseases worldwide (
1,
2). Selective serotonin reuptake inhibitors (SSRIs), despite their limitations, are the cornerstone of modern anxiolytic and antidepressant pharmacotherapy (
3). According to previous clinical and in vitro reports, escitalopram is the most selective (
4,
5) and is among the most efficient of SSRIs in the therapy of depression (
6,
7). However, therapeutic failure caused by a lack of response or by side effects in a significant portion of treated patients limits the clinical usefulness of escitalopram (
8). The high interindividual variation in escitalopram metabolism can mean either very high or very low escitalopram serum levels, potentially increasing the incidence of side effects or leading to insufficient clinical response and subsequently resulting in therapeutic failure (
9).
The initial biotransformation of escitalopram is predominantly catalyzed by the hepatic cytochrome P450 (CYP) enzyme CYP2C19 (
10,
11). The
CYP2C19 gene is highly polymorphic, and its genotypes determine the CYP2C19 enzymatic capacity;
CYP2C19*2 is the main loss-of-function (Null) allele (
12), while
CYP2C19*17 is thus far the only discovered gain-of-function allele (
13). Over the past decade, many studies explored the effect of
CYP2C19 genetic polymorphism on escitalopram exposure and treatment success. However, these studies have produced conflicting findings and failed to determine reliably whether
CYP2C19 genotype is a clinically relevant feature in escitalopram therapy. Some studies were performed in controlled settings but did not include a sufficient number of patients and therefore did not have the power to determine or quantify the consequences of
CYP2C19 variant alleles (
14,
15). Other studies included more patients but were performed under flexible-dosing protocols and without escitalopram concentration monitoring (
8,
16). In such settings, it is difficult to evaluate the effect of
CYP2C19 polymorphism on the therapeutic success of escitalopram. Consequently, unequivocal evidence is lacking for the clinical utility of
CYP2C19 genotyping in escitalopram-treated patients. Thus, in most psychiatric clinics worldwide, escitalopram therapy usually starts with the standard recommended dosage of 10 mg/day (
9), regardless of
CYP2C19 genotype.
Results
Escitalopram serum concentration was determined in 2,087
CYP2C19-genotyped patients 10 to 30 hours after drug intake.
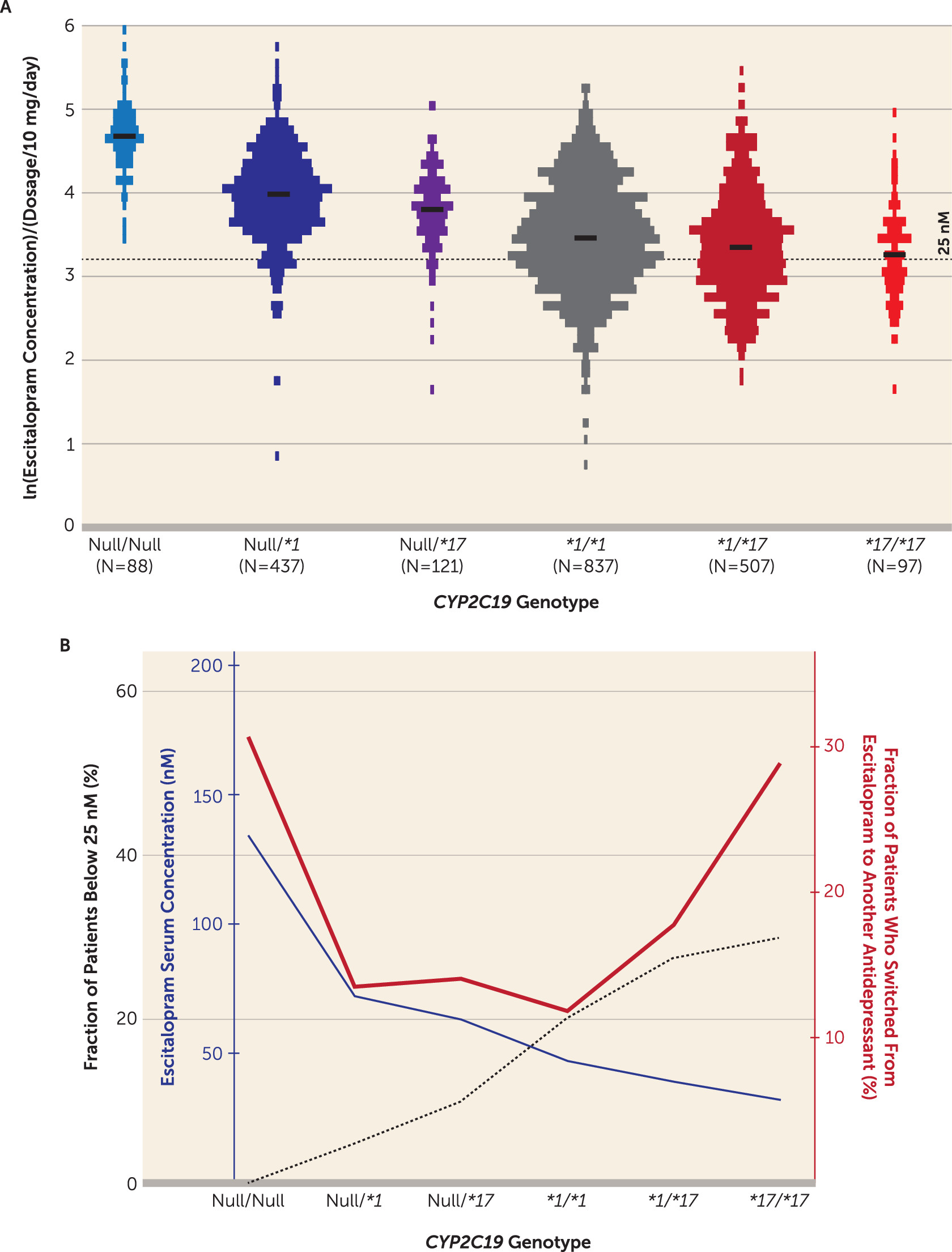
Figure 1A illustrates the dosage-harmonized serum concentrations of escitalopram according to the predicted order of CYP2C19 metabolic capacity, i.e.,
CYP2C19Null/Null,
CYP2C19*1/Null,
CYP2C19*17/Null,
CYP2C19*1/*1,
CYP2C19*1/*17, and
CYP2C19*17/*17.
To account for the variable number of measurements per patient, the effect of
CYP2C19 genotype on escitalopram concentration was estimated by a multivariate mixed-model analysis with age and sampling time as covariates. The serum concentration of escitalopram was significantly different between the
CYP2C19 genotype-defined subgroups (
Table 1). Mean estimates for escitalopram serum concentration were significantly greater among the poor and intermediate metabolizers compared with the extensive metabolizers—3.3 times greater for the poor metabolizers and 1.6 times (Null
/*1) and 1.4 times (Null/
*17) greater for the intermediate metabolizers (p<0.001) (
Table 2). At the other end, mean escitalopram serum concentrations were significantly lower among the ultrarapid metabolizers, by approximately 10% (
CYP2C19*1/*17) (p<0.003) and 20% (
CYP2C19*17/*17) (p<0.002) compared with the extensive metabolizers (
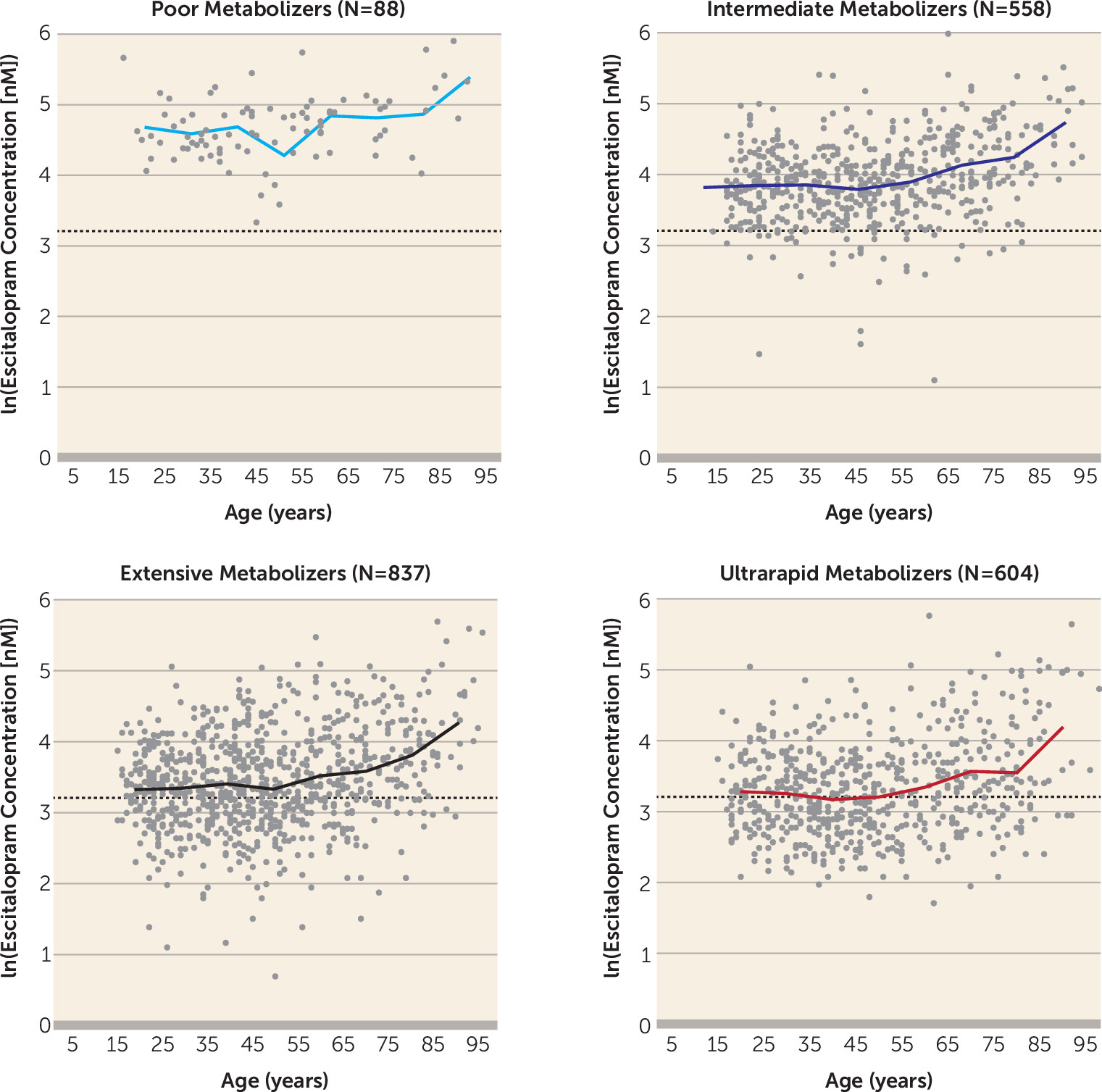
Table 2). In the mixed-model analysis, age and sampling time were found to be significant nongenetic covariates in explaining individual variability in escitalopram serum concentration. The dynamics of the increase in escitalopram exposure with age were similar regardless of
CYP2C19 genotype; the concentration remained fairly constant in patients under age 50, while a pronounced increase was observed in patients over age 65 (
Figure 2).
In the evaluation of individual measurements below the lower therapeutic boundary of escitalopram concentration defined for sufficient SERT inhibition (i.e., 25 nM), the proportion of patients with subtherapeutic levels increased with CYP2C19 metabolic capacity (
Table 2). Compared with the reference group (patients homozygous for
CYP2C19*1), which comprised 20.1% patients with subtherapeutic levels, the proportion of patients with escitalopram serum concentrations below 25 nM was significantly lower among the poor and intermediate metabolizers (p<0.001) and significantly higher among the ultrarapid metabolizers (p<0.003) (
Table 2). While none of the poor metabolizers had escitalopram serum concentrations below 25 nM after the integration of multiple concentration measurements, the odds ratio estimates for the incidence of concentrations below this boundary were 0.20 (
CYP2C19Null
/*1) and 0.44 (
CYP2C19Null
/*17) for the intermediate metabolizer subgroups and 1.5 (
CYP2C19*1/*17) and 1.7 (
CYP2C19*17/*17) for the ultrarapid metabolizers compared with the extensive metabolizers (
Table 2).
To address the effect of
CYP2C19 genotype on the probability of escitalopram therapeutic failure in naturalistic settings, the fraction of patients who switched to another antidepressant within 1 year after the last TDM analysis of escitalopram was compared between the subgroups. In the extensive metabolizer reference group, the 1-year switch frequency was 11.8% (
Table 2). Both the poor metabolizers and the ultrarapid metabolizers showed significantly higher frequencies of switching from escitalopram to another antidepressant compared with the extensive metabolizers, with odds ratios of 3.3 (p<0.001) for the poor metabolizers, 1.6 (p=0.003) for the
CYP2C19*1/*17 subgroup, and 3.0 (p<0.001) for the
CYP2C19*17/*17 subgroup. Regardless of
CYP2C19 genotype, escitalopram was most often switched to venlafaxine (34% of the overall cases), a combined serotonin-norepinephrine reuptake inhibitor, followed by switch to another SSRI (25%).
The effect of
CYP2C19 genotype on the various endpoints is depicted in
Figure 1B, which illustrates the relationship between relative exposure and proportion with therapeutic failure, by subgroup. Interestingly, the fraction of patients who switched to another antidepressant within 1 year formed a U-shaped curve, with higher rates of drug switch among the patients with the lowest and highest metabolic capacity.
Discussion
The large number of patients included in this study allowed quantification of the effect of CYP2C19 genotype on escitalopram exposure and therapeutic failure, as monitored by drug switching, at a relatively high statistical power. The results provide strong evidence that CYP2C19 genotype has a major clinical impact on escitalopram therapy through a profound effect on escitalopram exposure. In particular, CYP2C19 genotype–defined ultrarapid metabolizers and poor metabolizers were linked with an elevated risk of therapeutic failure. These findings suggest that preemptive CYP2C19 genotyping could prospectively be of value for the individualization of escitalopram dosing, resulting in increased drug efficacy and fewer cases of drug switching.
According to the genotype-associated escitalopram concentrations, a high proportion of the ultrarapid metabolizers exhibited escitalopram serum levels below the defined lower limit of effective concentration at the standard dosing regimen (10 mg/day). This is the likely explanation for the higher chances of therapeutic failure in this subgroup, since the chronic effect of continuous lower levels of escitalopram, for weeks or months, causing insufficient receptor occupancy, will have a higher phenotypic effect than anticipated just from the serum levels. The necessity of achieving an adequate escitalopram serum concentration quickly is of particular relevance for certain patient subpopulations, such as those with high rates of nonadherence or with suicidal thoughts. According to our results,
CYP2C19 genotype–defined ultrarapid metabolizers exhibit an increased incidence of therapeutic failure and exposure to subtherapeutic escitalopram serum concentrations. This may be related to our previous results (
22) showing higher suicidality among suicide attempters who are ultrarapid CYP2C19 metabolizers as compared with poor metabolizers, intermediate metabolizers, or extensive metabolizers, based on a study of 207 depressed patients.
Poor metabolizers exhibited a 3.3-fold increase in drug concentrations compared with extensive metabolizers, caused by the absence of CYP2C19-mediated escitalopram metabolism. The effect on exposure was paralleled by a higher incidence of drug switching in poor metabolizers compared with extensive metabolizers. Well-replicated results have shown that the occurrence and severity of escitalopram-induced side effects such as insomnia, diarrhea, somnolence, dizziness, increased sweating, constipation, fatigue, and indigestion are dose dependent (
23–
26) and occur two to three times more frequently in patients receiving 20 mg/day compared with those receiving 10 mg/day. Therefore, the likely explanation for the higher frequency of therapeutic failure in poor metabolizers is an increased risk of dose-dependent side effects due to elevated escitalopram serum levels. Thus, the results from this study suggest that
CYP2C19 genotype–guided dosing may have the potential to improve the clinical success of escitalopram therapy and could be considered as a possibility in routine clinical practice. Ultrarapid and poor metabolizers represent the subpopulations that would benefit the most from a successful individualization of escitalopram dosing based on
CYP2C19 genotype. Ultrarapid and poor metabolizers together represent 33% of our study population. Considering that 16 million people in the United States (
27) and more than 300 million people in the world (
28) suffer from depression, preemptive genotyping for
CYP2C19 could then be expected to increase the success rate of escitalopram-based antidepressant therapy for millions of patients around the world, and to a greater extent in countries with high prevalences of
CYP2C19*2 and
CYP2C19*17, notably, for example, in East and South Asia, where the frequency of
CYP2C19*2 exceeds 30% (
17).
The Clinical Pharmacogenetics Implementation Consortium (CPIC) (
9) recommends 1) initiating therapy at the standard recommended starting dosage of escitalopram (10 mg/day) for intermediate metabolizers and extensive metabolizers; 2) considering either reducing the dosage of escitalopram by 50% or selecting an alternative drug not predominantly metabolized by CYP2C19 for poor metabolizers; and 3) selecting an alternative drug not predominantly metabolized by CYP2C19 for ultrarapid metabolizers. The data presented here suggest the need for further discussion on the escitalopram dosing recommendations. According to our exposure-by-genotype data in this study, it appears that 5 mg/day is generally sufficient to reach therapeutic serum levels in poor metabolizers, whereas for intermediate and extensive metabolizers, the standard dosage (10 mg/day) is more appropriate, as suggested by the CPIC guidelines. Young ultrarapid metabolizers appear to be a group for whom a dosage increase from 10 mg/day to 20 mg/day is frequently necessary to reach sufficient serum levels of escitalopram (
Figure 1 and
Figure 2). Of course, in certain cases, TDM can be utilized to support decisions on whether to further modify escitalopram dosing, and it is of particular importance in patients with hepatic or renal functional impairments and patients on polypharmacy. Escitalopram exposure increases in the older ages, unrelated to
CYP2C19 genotype, an effect that is likely due to an age-dependent decrease in hepatic or renal perfusion. The data presented here suggest that patients over age 65 are not likely to benefit from dosages above 10 mg/day even if they are in the ultrarapid metabolizer subgroup, which is in agreement with the U.S. Food and Drug Administration drug label for escitalopram (
29).
Several limitations of this study are worth mentioning. One is the unavailability of data that could be used to estimate the patients’ volumes of distribution. Since the serum concentration of a drug at a specific time point is inversely proportional to the volume of distribution, it is likely that including this information as a covariate would significantly improve the estimate of the CYP2C19 genotype effects on escitalopram serum concentration. Information about comedication with drugs that potentially create pharmacokinetic interactions is another factor of importance for the variability in serum concentration of escitalopram. Lack of data on comedication therefore represents a study limitation, in line with other nongenetic factors that may also affect drug concentrations (e.g., comorbidity).
Second, although the available information on switching to other antidepressants can be used as a clear indication of therapeutic failure, the exact information on diagnosis and treatment outcome at the time of blood sampling cannot be obtained. Consequently, a follow-up hypothesis-oriented clinical study in more controlled settings is needed to evaluate the extent of the benefit of using CYP2C19 genotype to tailor escitalopram therapy.
Finally, despite all the data indicating that preemptive
CYP2C19 genotyping can be useful in escitalopram therapy, the cost-effectiveness of such an intervention, which is of relevance for routine usage and reimbursement, is beyond the scope of this report. A previous study demonstrated the cost-effectiveness of preemptive
CYP2C19/CYP2D6 genotyping in antipsychotic therapy (
30), and a similar study is needed to assess the cost-benefit ratio of
CYP2C19 genotyping in escitalopram therapy.