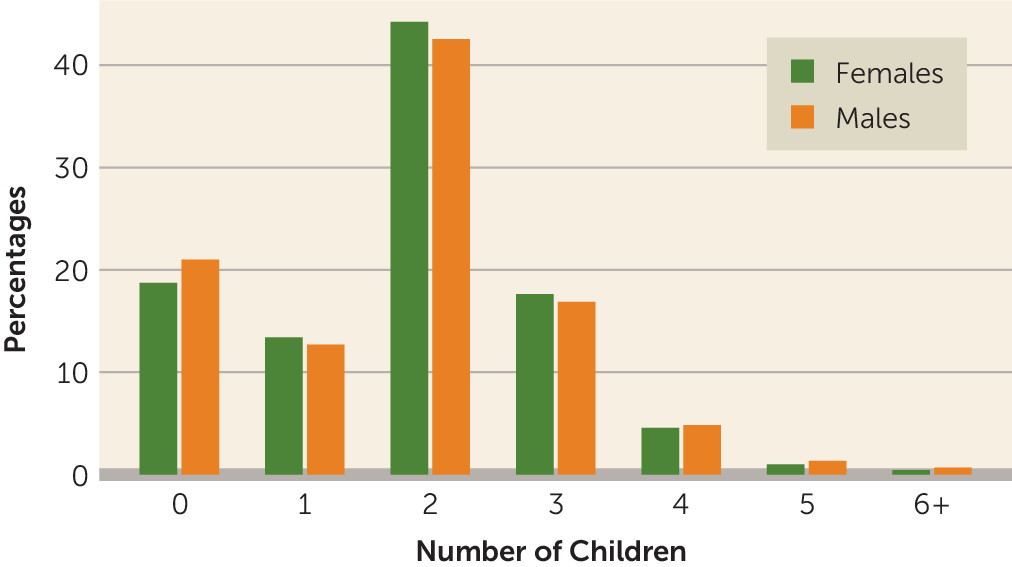Given improvements in medical care as well as the reduction in prenatal, infant, and child mortality in developed countries, the number of offspring can be considered a measure of an individual’s reproductive fitness (
1). Some studies have reported that people with schizophrenia, particularly males, have substantially fewer children compared with individuals in the general population (
2–
4). However, the relationship between an individual and the number of his or her offspring is a complex phenotype. In addition to variance in aspects of fundamental biology related to fertility and fetal survival, the number of offspring is also driven by an individual’s mating choices and opportunities, reflecting the individual’s social, cultural, religious, economic, and historical environment. It is unclear which of these variables is primarily responsible for the association between schizophrenia and low fecundity.
Schizophrenia has a substantial heritability (
5,
6), to which alleles across the frequency spectrum contribute (
7–
9). As expected for a disorder associated with low fecundity, alleles that contribute large effects on risk are rare, with their frequencies being dictated by the balance between the rates of new (de novo) mutation formation and removal of these mutations by strong negative selection, which typically occurs within a few generations (
10). However, a substantial component of liability to schizophrenia is conferred by risk alleles that are common and that individually confer small effects on risk (
11,
12). There has been a long-standing debate, from an evolutionary perspective, as to how common risk alleles persist in the population (
13). Proposed hypotheses include that schizophrenia alleles confer reproductive advantages in unaffected people, depending on their particular genetic background or environmental context (historical or contemporary), that risk alleles are maintained by linkage to positively selected alleles, that common risk alleles for schizophrenia are individually too weak to be effectively purged by negative selection, and that risk alleles occur in regions where the effects of more deleterious variants cause haplotypes to be removed from the gene pool, which reduces genetic diversity, impairs the efficiency of selection, and allows alleles with small deleterious effects to increase in frequency by drift (
13). Of these possible explanations, perhaps the most widely known is the notion that schizophrenia may persist in modern populations because individuals who carry risk alleles but who do not manifest schizophrenia have psychological traits that increase their fecundity. For example, Jarvik and Deckard (
14) proposed that schizoid-paranoid personality traits found in nonpsychotic relatives of individuals with schizophrenia (which they termed the Odyssean personality) may be advantageous in a world “plagued by terror, strife, and war.”
In a recent study on reproductive fitness and genetic risk of psychiatric disorders in 93,720 genotyped Icelanders (≥45 years old, without a diagnosis of a psychiatric disorder) (
15), the authors found no evidence that common schizophrenia risk alleles confer reproductive advantage, although, as expected, rare alleles with large effect sizes in the form of copy number variants did confer reproductive disadvantage, with a greater fecundity reduction in males than females.
In this study, we sought to examine the relationship between schizophrenia liability and fecundity using the UK Biobank (
16), a powerful resource for testing hypotheses at the population level. Specifically, we sought to test the hypothesis that the burden of common risk alleles for schizophrenia is associated with reproductive advantage in individuals without schizophrenia. Given the sex-related effects of the disorder on fecundity, we also tested male and female risk allele carriers separately.
Methods
UK Biobank Data
The UK Biobank comprises a large prospective cohort of more than half a million residents of the United Kingdom for whom data on genetics and number of children are available. The phenotype “number of children” is the self-reported response to the following question: “How many children have you fathered (or mothered)?” If the answer was >200, then it was discounted by the UK Biobank. For responses >15, the participant was asked to confirm the answer.
The schizophrenia genome-wide association study (GWAS) cohort that we used to define risk alleles was primarily of European ancestry, and thus we restricted the sample to individuals who self-reported as being Caucasian from the United Kingdom or of Irish ancestry (N=140,494). In the first wave of UK Biobank data, 227 people (79 males, 148 females) had self-reported schizophrenia or had hospital admissions for an episode of schizophrenia or schizoaffective disorder. These individuals were excluded because we were interested in examining people who did not express the disorder. We also excluded 558 individuals with missing data on number of children. Thus, 139,679 participants were included in our analyses.
For the UK Biobank, informed consent was obtained from all participants, and the present study was conducted under generic approval from the NHS National Research Ethics Service (
16).
Measuring Burden of Risk Alleles: Polygenic Risk Score (PRS)
To define risk alleles, we used the largest available schizophrenia GWAS comprising a meta-analysis of two large studies: the most recent study conducted by the Psychiatric Genetic Consortium (PGC-2) (
8) and the most recent study of individuals with a clinical diagnosis of schizophrenia attending a clozapine clinic compared with control subjects (CLOZUK) (
13). The schizophrenia data sets were imputed using the SHAPEIT and IMPUTE2 software programs (
17,
18), with a combination of the 1000 Genomes phase 3 (1KGPp3) and UK10K data sets as a reference panel. Data were available for a total of 40,675 schizophrenia case subjects and 64,643 control subjects (
13). For constructing PRS in the UK Biobank, we included autosomal imputed genetic data that passed stringent quality control criteria (minor allele frequencies ≥0.01) and an imputation quality score ≥0.9. This resulted in 5,471,613 single-nucleotide polymorphisms (SNPs). Using the UK Biobank genotypes, we performed clumped pruning by using the parameters (r
2) of 0.2, a physical distance threshold for clumping SNPs of 1 Mb, and an association p value threshold of 0.05 in the GWAS of PGC2 and CLOZUK. The clumped pruning process retains SNPs that are the most significantly associated with schizophrenia while excluding SNPs at which the genotypes are correlated with the selected SNPs.
We selected markers on the basis of the schizophrenia association significance threshold (p<0.05) to construct a polygenic score in the UK Biobank data (markers, N=32,576). A p value <0.05 is the threshold that maximally captures polygenic risk in current studies of schizophrenia (
8). The PRS was calculated from the effect size-weighted sum of associated alleles within each study subject. PRSs were adjusted for array (two different arrays were used in the UK Biobank) and the first eight principal components and then standardized by subtracting the population mean for PRS and dividing by the standard deviation. The first eight principal components, of 15 available in the biobank, were selected after visual inspection of each pair of principal components, taking forward only those that resulted in multiple clusters of individuals (
19).
Statistical Analysis
We used Poisson regression analysis to test number of children for association with schizophrenia PRS, covarying for age and sex. (Age was strongly positively associated with number of children in all analyses.) We examined the interaction between sex and schizophrenia PRS by comparing two Poisson regression models: PRS, age, and sex and PRS, age, sex, and PRS-by-sex interaction. Because the distributions of the numbers of children born to males and females may differ (
15), we also performed this analysis for males and females separately.
As a test of nonlinear effects, we tested seven groups according to number of children (0, 1, 2, 3, 4, 5, ≥6) and performed a logistic regression analysis (adjusted for principal components, array, and age as described above), examining whether childless participants had a greater or lower genetic liability to schizophrenia compared with participants with a particular number of children. We used the truncated six-children measure for the interaction analysis, because the number of UK Biobank participants who reported more than six children was small (N=288), and the inclusion of multiple small groups may have adversely affected the robustness of the analyses.
We report B coefficients for Poisson regression analyses and odds ratios (odds ratio=exponential[B]) for the logistic regression analyses.
To compare the effect of the PRS on fecundity among individuals unaffected by schizophrenia with the effects observed among participants with schizophrenia, we used Fisher’s fundamental theorem of natural selection (
20,
21), which establishes a relationship between fecundity and strength of selective pressure based on additive genetic variance. The set of calculations we performed is detailed in the
online supplement. Briefly, we retrieved data from GWASs and epidemiological studies and used the data to calculate the variance in fecundity in the general population (including among individuals with schizophrenia) explained by the PRS. Then we compared the value with the variance in fecundity that, according to our data, could be explained by the PRS for unaffected individuals. The ratio of both variances allowed us to compare the strength of selective pressures (i.e., negative selection in affected individuals compared with positive selection in unaffected individuals) and infer their effect in the general population.
Results
PRS and Number of Children
For males and females without a diagnosis of schizophrenia combined, number of children was weakly but significantly positively associated with schizophrenia PRS (B=0.006, 95% CI=0.002, 0.010, p=0.0017) (
Table 1). The effect size was similar to one previously reported for an Icelandic population (
15) (B=0.006, p=0.160), although in that study, the association was not statistically significant. The quadratic test for nonlinear relationships was not significant in both studies.
Fecundity Effects in Healthy and Affected Individuals
The observed positive association between schizophrenia PRS and fecundity applies to the subset of the general population that is unaffected by schizophrenia. When this effect is extrapolated to the general population, it can be compared with the known negative association between fecundity and onset of schizophrenia (
2–
4). After both effects are considered, in light of the population prevalence of schizophrenia, the negative effect contributed by the disorder to the population is substantially greater than the positive effect associated with the PRS (twofold to 15-fold) (for further details, see the
online supplement).
PRS and Number of Children Among Males and Females Separately
The percentages of males and females in each category of number of children are presented in
Figure 1. Polygenic risk of schizophrenia was not associated with number of offspring among males (B=0.001, 95% CI=–0.005, 0.007, p=0.745), but it was positively associated with number of offspring among females (B=0.011, 95% CI=0.006, 0.016, p=7.1×10
−5) (
Table 1).
We further explored the patterns of association among males and females, comparing the PRS among persons with specific numbers of offspring (
Tables 2 and
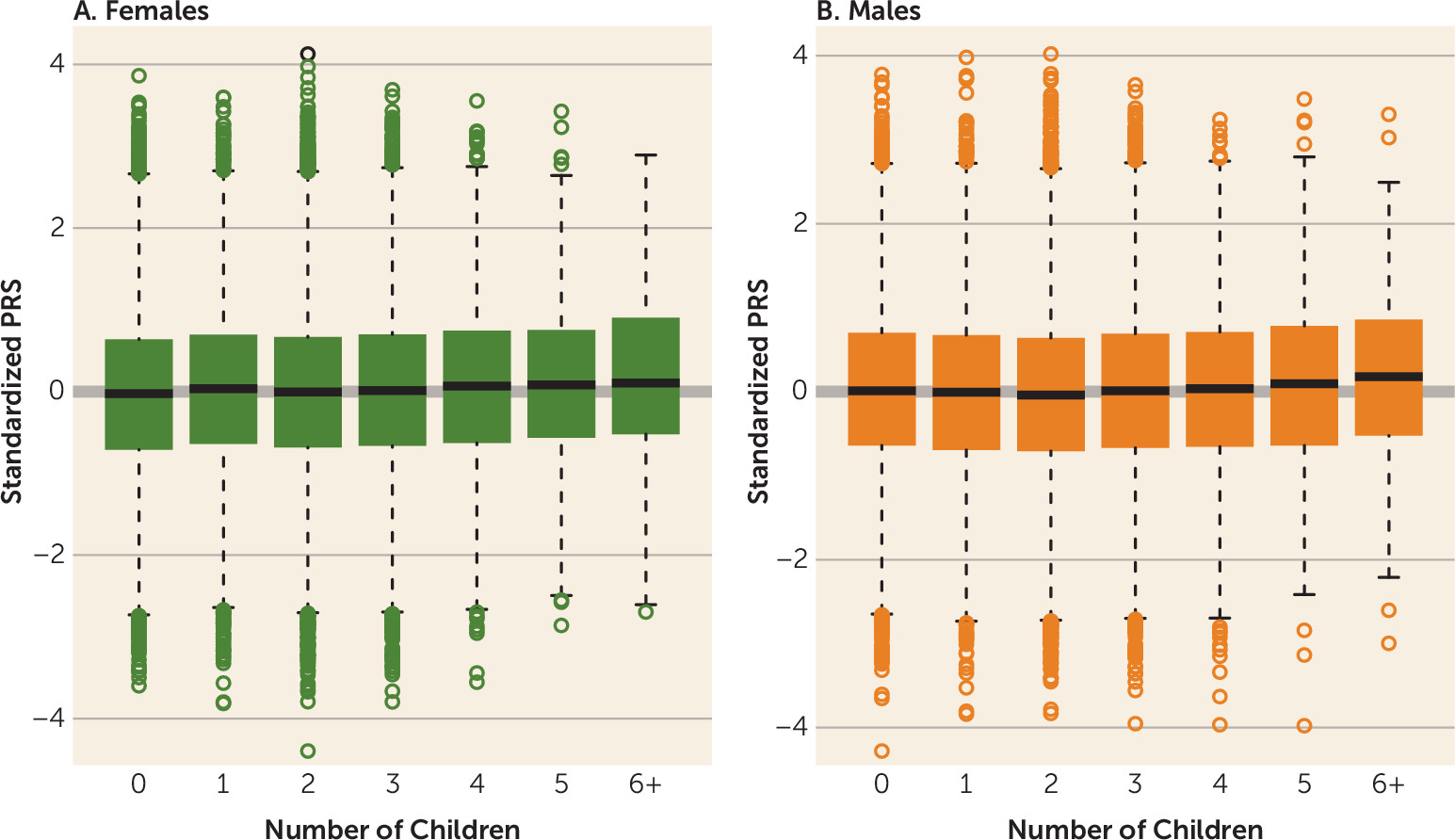
3) with the PRS among those without children. The odds ratio was derived as the exponential of the logistic regression beta coefficient estimate and represents the change in the odds of inclusion in the target group, defined by the exact number of children, with an increase of one standard deviation in PRS. The distribution of the PRS among females and males by number of children is shown in
Figure 2. Across all categories for number of children, PRS was positively associated with number of offspring among females; that is, the higher the liability to schizophrenia, the more children they have (
Table 2,
Figure 2A). In the male cohort, the effect was less consistent. Males who fathered one or two children had lower schizophrenia PRSs compared with those with no children. Indeed, schizophrenia PRS was associated with an absence of children among men (odds ratio=0.967, 95% CI=0.94, 0.99, p=0.015 and odds ratio=0.942, 95% CI=0.92, 0.96, p=2.2×10
−8) compared with the PRSs of men with one and two children, respectively, in this cohort (
Table 3,
Figure 2B). (In
Table 3 the odds ratios were derived as the exponential of the logistic regression Beta coefficient estimate and represent the change in the odds of membership in the target group, defined by the exact numbers of children with every increase of one standard deviation in PRS.) Moreover, schizophrenia PRS was elevated among those with a greater number of offspring (≥5), although the association fell short of statistical significance. In comparing PRSs among men with and without children, higher genetic liability to schizophrenia was associated with no offspring (odds ratio=0.96, 95% CI=0.94, 0.98, p=8.01×10
−5).
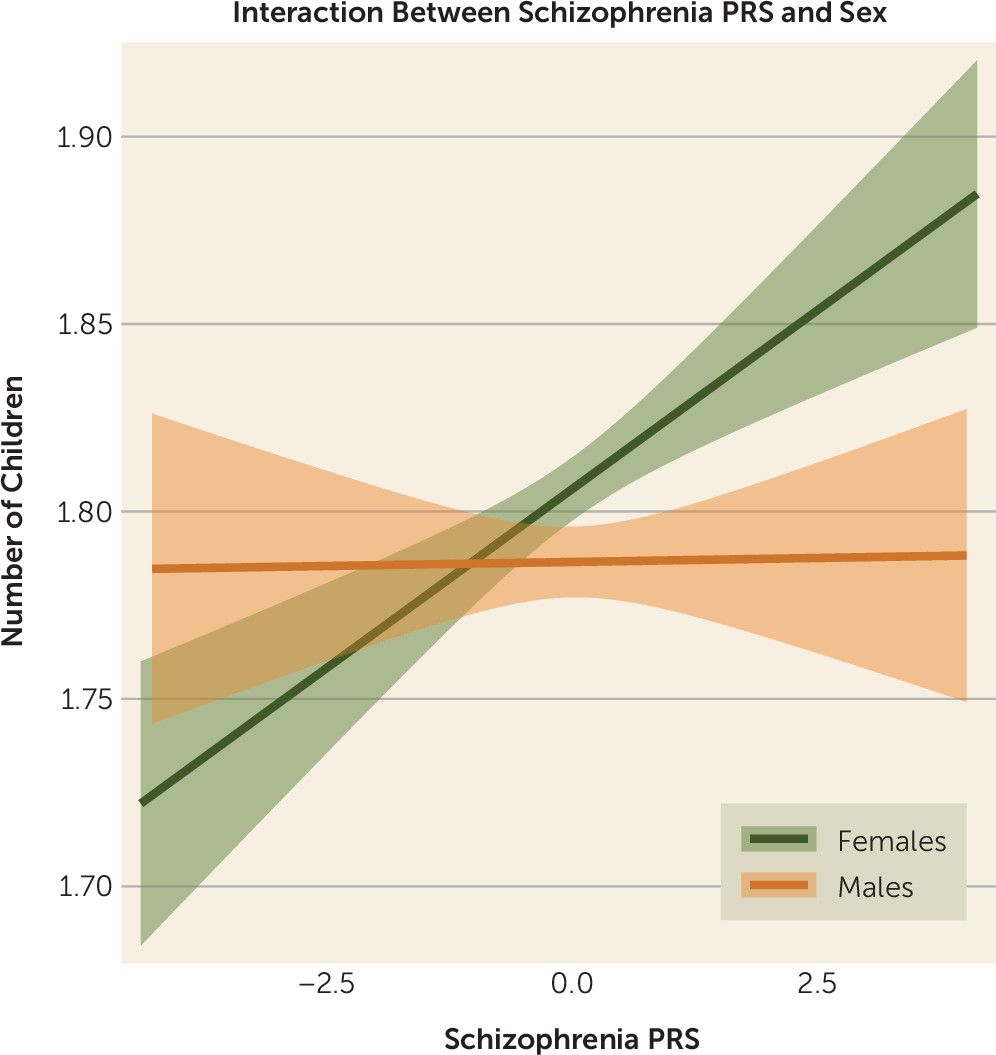
PRS-by-Sex Interaction
We tested for an interaction effect between number of offspring, liability to schizophrenia (PRS), and sex using Poisson regression. The outcome variable was number of children, and the predictors were schizophrenia PRS, sex, and an interaction term (schizophrenia PRS by sex). The interaction term was significant (B=−0.011, SE=0.004, p=0.009) over and above schizophrenia PRS (B=0.01, SE=0.003, p=8.8×10
−5), sex (B=−0.021, SE=0.004, p=2.9×10
−7), and age (B=0.015, SE=0.0003, p<10
−16), respectively. The negative sign of the interaction term B coefficient indicated that the increase in the number of children among women with higher genetic loading to schizophrenia was greater than that among men (in the UK Biobank, women were coded as “0,” and men were coded as “1”) and was more pronounced when we compared persons with and without offspring (B=−0.08, SE=0.014, p=5.5×10
−10) using logistic regression. The interaction of sex and PRS on number of offspring is illustrated in
Figure 3.
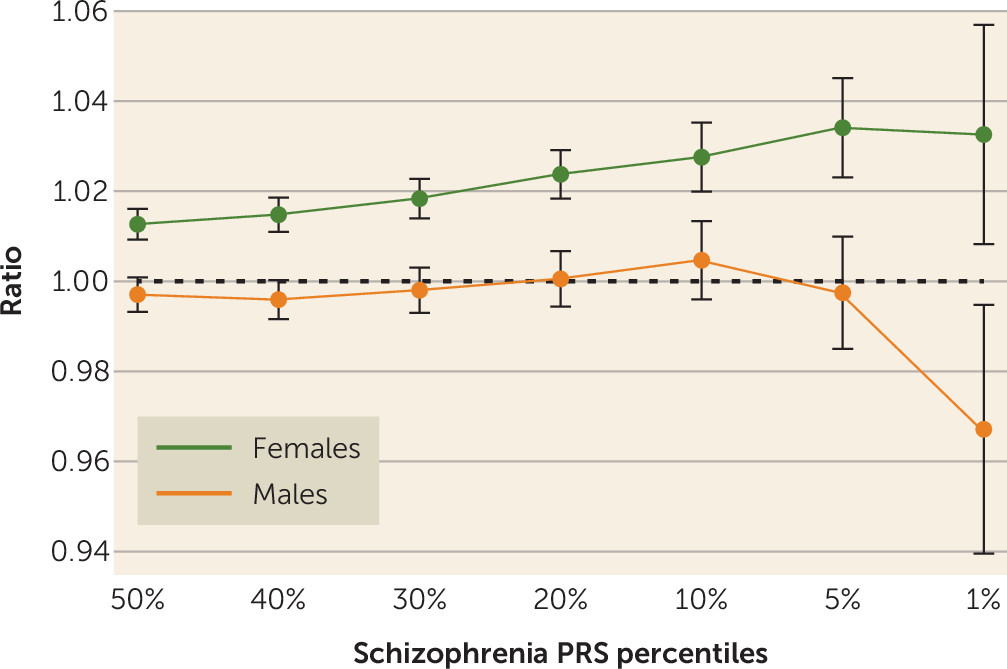
Given the nonlinear relationship between PRS and fecundity in males (
Table 3), we further explored the different patterns of relationships between genetic liability to schizophrenia and the total numbers of children among males and females. We report these as ratios of the mean numbers of offspring born to males and females in the 50th, 40th, 30th, 20th, 10th, 5th, and 1st percentiles of the schizophrenia PRS distribution to the total population mean (
Figure 4). Relatively more children were born to females with higher PRS, and this relationship increased toward the PRS extreme, plateauing at the 1st percentile. Among men, a more complex pattern was observed, with an initial decrease in the relative fecundity as PRS increased, followed by an increase and then, at the extreme end of the distribution, a sharp decrease in relative fecundity.
Discussion
We sought to test the hypothesis that the burden of common risk alleles for schizophrenia is associated with reproductive advantage in individuals without schizophrenia and that this may explain the persistence of common risk alleles in the population in the face of negative selection among persons affected by schizophrenia. Our findings suggest that at a population level, higher common variant genetic loading to schizophrenia is associated with a small reproductive advantage among females. The effect was not observed among males, and there is evidence that persons with the highest risk scores have fewer offspring. When males and females were considered together, there was a net reproductive advantage associated with a high PRS. However, the effects of PRS are not sufficient to explain the persistence and allele frequencies of schizophrenia risk alleles in contemporary populations, because these effects are small relative to the effects of schizophrenia itself on fecundity, which at a population level have a stronger impact.
In considering the effects of PRS that we have observed in unaffected individuals, we should bear in mind that schizophrenia liability is highly pleiotropic (
22); that is, alleles that increase risk for schizophrenia also increase liability to other clinical, cognitive, and behavioral traits, some of which may be associated with higher fecundity. For example, people with relatively poorer cognitive ability, a phenotype that is associated with higher schizophrenia polygenic risk (
23), may be less motivated, have less opportunity, or have less ability to control fertility. It is important to emphasize that links between fecundity on the one hand and behavioral and cognitive traits on the other are highly dependent on the contemporary environmental context and that the present results apply to the population in the United Kingdom, which is among the wealthiest in the world. Whether these results generalize globally and historically will depend on whether the effects are mediated primarily by direct effects on biology or, as we speculated above, by an interplay between behavioral and personal traits and social, cultural, religious, and economic norms that are highly variable across history and across societies. This hypothesis therefore will have to be tested in different countries and cultures.
It has previously been documented that the effect of schizophrenia on fecundity is more pronounced among males (
2,
4,
24). Our population-based study demonstrated that higher genetic liability to schizophrenia was associated with childlessness among males. We also showed a significant statistical interaction between sex and genetic liability to schizophrenia. Men at the extreme end of the liability scale of the PRS distribution (the 1st percentile) had fewer children compared with men in other PRS percentiles (
Figure 4).
The sex-related differences between number of children and schizophrenia liability is consistent with a family-based study (
25) in which the female siblings of people with schizophrenia had a significantly greater number of children, while the male siblings of affected individuals had significantly fewer children. This finding as well as the findings in the present study are consistent with the idea of sexual selection (
26), whereby the number of offspring is substantially the result of female choice. Thus, the association between high PRS and childlessness among males (but not females) is consistent with female mating choices favoring males with characteristics associated with lower schizophrenia liability (for example, one might speculate better social cognition), whereas male mating choice is less relevant. However, other explanations are possible. Schizophrenia liability may differentially influence the reproductive biological fitness of males and females, or behavioral and cognitive traits that would be selected against by both sexes may be differentially affected by schizophrenia liability in males and females.
Strengths of our study are the use of the largest European ancestry schizophrenia data set to date to identify genetic risk loci and the largest available genotyped population cohorts to generate schizophrenia PRSs as well as the ability to measure liability to schizophrenia directly at a molecular level. This enabled us to test the hypothesis with extremely high power, including testing the effects on males and females separately. There are three limitations of our study. First, it was based on common SNPs, and the effects on fecundity of rare variants were not tested. Second, we were unable to test mechanisms (i.e., whether the genetic effects were mediated by behavior or by reproductive biology). Lastly, the conclusions are specific to place (the United Kingdom) and time (participants largely of reproductive age in the latter half of the 20th century).
Acknowledgments
Dr. Walters is supported by a JMAS Sim Fellowship from the Royal College of Physicians of Edinburgh. This study was conducted using the UK Biobank resource. UK Biobank was established by the Wellcome Trust, Medical Research Council, Department of Health, Scottish Government, and Northwest Regional Development Agency. UK Biobank has received funding from the Welsh Assembly Government and the British Heart Foundation. Data collection was funded by UK Biobank. The work at Cardiff University was supported by Medical Research Council (MRC) Centre (grant MR/L010305/1) and by a program grant (grant G0800509). The CLOZUK sample was genotyped with funding from the European Union’s Seventh Framework Programme for research, technological development, and demonstration (grant 279227) (CRESTAR Consortium).