Current diagnostic criteria and symptom severity measures rely on the nine core symptoms of major depressive disorder (
1–
3), yet impairments as a result of major depression and improvements with treatment are not fully captured by these symptom criteria (
4–
7). Irritability is unique among major depressive disorder-associated symptom domains, because it is considered to be a core diagnostic symptom among adolescents but not adults (
1). Yet, it is widely prevalent in adult patients with major depression, with 40%−50% reporting the presence of irritability for more than half the time in their current depressive episode (
8–
10). The presence of irritability is associated with greater severity of depressive and anxiety symptoms, earlier age at onset, presence of atypical features, and poorer quality of life (
8,
9,
11). Irritability is also associated with poorer clinical course. Patients with major depression who report irritability, compared with those without irritability, are more likely to have a chronic course characterized by a greater number of weeks spent in a depressive episode and greater time spent with residual symptoms in between depressive episodes (
10). The presence of anger or hostility before treatment initiation and worsening of irritability after initiation of antidepressant medication are both associated with lower acute-phase remission rates (
12,
13). Reductions ≥30% in anger or hostility ratings by week 2 of antidepressant treatment are associated with a doubled likelihood of remission among patients with major depression (
14). In patients with treatment-resistant major depression, the presence of irritability may help guide the optimal next-step treatment selection, as evidenced by greater symptom improvement with low-dose brexpiprazole in patients with irritability (
15).
Despite the wide prevalence, the prognostic utility, and the treatment selection potential, measurement-based care protocols (
16,
17) and treatment guidelines (
18,
19) for major depressive disorder do not systematically assess or incorporate irritability in clinical decision making. To be clinically useful, changes in irritability should predict longer-term clinical outcomes and reflect improvement beyond what is reflected in measures of overall depressive symptom severity. Previous studies have not evaluated whether changes in irritability with antidepressant treatment were completely accounted for by changes in depressive symptom severity. Furthermore, the prognostic significance of changes in irritability has not been shown to be independent of reduction in depressive symptom severity. Finally, even if changes in irritability predicted longer-term outcomes, currently there are no easy-to-use methods or recommendations to incorporate them in clinical decision making at the individual patient level.
Our aim in this study was to evaluate the clinical utility of adding irritability to the current paradigm of measuring depressive symptom severity during the course of antidepressant treatment. The specific questions we asked, using two samples of convenience, were as follows:
1.
Does irritability improve from baseline to week 4 of antidepressant treatment even after accounting for change in depressive symptom severity?
2.
Does baseline-to-week-4 change in irritability predict remission and no meaningful benefit (<30% reduction from baseline) at week 8 even after controlling for baseline irritability, baseline depressive symptom severity, and baseline-to-week-4 change in depressive symptom severity?
3.
Can baseline-to-week-4 changes in irritability and depressive symptom severity be used to predict remission and no meaningful benefit at an individual level?
4.
Do these predictions replicate in a separate, unrelated sample of outpatients with major depression?
Using the first sample of participants (N=664) from the Combining Medications to Enhance Depression Outcomes (CO-MED) trial, baseline-to-week-4 change in irritability was assessed after controlling for change in depressive symptom severity. Prediction of acute-phase (week 8) treatment outcomes (remission and no meaningful benefit) by baseline-to-week-4 change in irritability was tested in models incorporating baseline irritability and depressive symptom severity and baseline-to-week-4 change in depressive symptom severity. The estimates obtained from the CO-MED trial were used to predict individual-level probability of remission and no meaningful benefit in a second sample of participants (N=163) from the Suicide Assessment Methodology Study (SAMS). Week 4 was selected to measure change from baseline in irritability and depressive symptom severity because it is the first critical decision point for assessing response to treatment per measurement-based care protocol (
16). Because SAMS was an 8-week study, acute-phase treatment outcomes were ascribed at week 8. Although remission (defined as a score ≤5 on the Quick Inventory of Depressive Symptomatology–Clinician-Rated [QIDS-C] [
3]) is the preferred goal of acute-phase antidepressant treatment (
19), “no meaningful benefit” (defined as a reduction <30% in QIDS-C score from baseline) was included to facilitate clinical decision making (treatment augmentation, switching, or discontinuation).
Methods
Study Overview and Participants
CO-MED trial.
Our analysis included all CO-MED trial (NCT00590863) participants for whom data from the Concise Associated Symptom Tracking (CAST) scale (
20) were available at baseline (N=664). The details of the CO-MED trial, including recruiting sites, inclusion and exclusion criteria, and institutional review board approvals, have been described previously (
21). From March 2008 through February 2009, participants from six primary care sites and nine psychiatric care sites were enrolled after written informed consent was obtained (
21). Inclusion was restricted to treatment-seeking outpatients with major depression who were 18–75 years old and had nonpsychotic chronic (duration of current episode ≥2 years) or recurrent (current episode ≥2 months) depression and a baseline score ≥16 on the 17-item Hamilton Depression Rating Scale (HAM-D) (
22). Exclusionary criteria included a lifetime history of a psychotic disorder, current psychotic symptoms, a history of an eating disorder in the past 2 years, a current primary diagnosis of obsessive-compulsive disorder (OCD), current substance dependence requiring inpatient-level care, an unstable general medical condition, a current psychiatric condition necessitating hospitalization, a history of a seizure disorder or narrow-angle glaucoma, inadequately treated hypothyroidism, and use of contraindicated medications for general medical or psychiatric conditions (antipsychotics, anticonvulsants, mood stabilizers, CNS stimulants, antidepressants, or other medications with potential augmentation properties). Participants were randomly assigned at baseline to one of three treatment arms in a 1:1:1 ratio after stratification by clinical site: escitalopram plus placebo (selective serotonin reuptake inhibitor [SSRI] monotherapy), sustained-release bupropion plus escitalopram (bupropion-SSRI combination), and extended-release venlafaxine plus mirtazapine (venlafaxine-mirtazapine combination). Postrandomization visits were conducted at weeks 1, 2, 4, 6, 8, 10, and 12 for the acute phase and weeks 16, 20, 24, and 28 for the continuation phase. As previously reported (
21), acute-phase and continuation-phase outcomes (remission or response) did not differ among the three treatment arms.
SAMS.
All SAMS (NCT00532103) participants who completed the QIDS-C and CAST assessments at baseline and at week 4 and the QIDS-C at week 8 (N=163) were included. The details of SAMS, including recruiting sites, inclusion and exclusion criteria, and institutional review board approvals, have been described previously (
23). From July 2007 through February 2008, a total of 266 participants from six primary care sites and nine psychiatric care sites were enrolled in SAMS after written informed consent was obtained (
23). Inclusion was restricted to 18- to 75-year-old treatment-seeking outpatients with nonpsychotic major depressive disorder and a score ≥14 on the HAM-D. Exclusion criteria included failure of two or more courses of SSRIs in the current episode, current substance use disorder, bipolar disorder, schizophrenia, a primary diagnosis of OCD or an eating disorder, use of prohibited medications (antipsychotics and antiepileptics), and a unstable general medical or psychiatric condition necessitating hospitalization.
Measurement-Based Care
The measurement-based care (
16) approach was used in both the CO-MED trial and SAMS to make medication dosage adjustments during the 8-week postbaseline period, using the QIDS-C (
3) as a measure of depression severity and the Frequency, Intensity, and Burden of Side Effects Rating Scale (
24) as a measure of side effects. Study physicians made dosage increases only if the depression severity was not adequately controlled and the side effect burden was tolerable.
Medications
CO-MED trial.
Participants in all three treatment arms received two types of pills in a single-blind fashion. The study personnel were aware of both pill types, but study participants were aware of only the first pill type. In the SSRI monotherapy arm, escitalopram was started at 10 mg/day, with an increase up to 20 mg/day permitted at week 4; pill placebo was added as the second pill type at week 2. In the bupropion-SSRI treatment arm, sustained-release bupropion was initiated at 150 mg/day and increased to 300 mg/day at week 1; escitalopram was started at 10 mg/day as the second pill type at week 2, and dosage increases of bupropion (up to 200 mg twice daily) and escitalopram (up to 20 mg/day) were permitted from weeks 4 to 8. In the venlafaxine-mirtazapine arm, extended-release venlafaxine was initiated at 37.5 mg/day and titrated to 150 mg/day by week 1; mirtazapine at 15 mg/day was added as the second pill type at week 2, and dosage increases of venlafaxine (up to 300 mg/day) and mirtazapine (up to 45 mg/day) were permitted from weeks 4 to 8.
SAMS.
Participants were treated with an SSRI (escitalopram, citalopram, sertraline, paroxetine, controlled-release paroxetine, or fluoxetine) in an open-label fashion, with choice of an antidepressant at the individual participant’s discretion (
20). Escitalopram was initiated at 10 mg/day, and an increase up to 20 mg/day was permitted from weeks 4 to 6. Citalopram was initiated at 20 mg/day, and an increase up to 40 mg/day was permitted from weeks 4 to 6. Sertraline was initiated at 50 mg/day and increased to 100 mg/day at week 2; further increases up to 150 mg/day were permitted from weeks 4 to 6. Paroxetine was initiated at 20 mg/day, and an increase up to 40 mg/day was permitted from weeks 4 to 6. Controlled-release paroxetine was initiated at 25 mg/day, and an increase up to 37.5 mg/day was permitted from weeks 4 to 6. Fluoxetine was initiated at 20 mg/day, and an increase up to 40 mg/day was permitted from weeks 4 to 6.
Assessments
QIDS-C.
The 16 items of the QIDS-C are based on the nine symptom criteria domains of major depressive disorder. Each item is scored from 0 to 3, and total score ranges from 0 to 27 (
3,
25). The QIDS-C correlates highly with the 17-item HAM-D (r=0.93) and has high inter-item correlations (Cronbach’s alpha=0.85) (
25,
26). Remission was defined as a score ≤5 on the QIDS-C at week 8. No meaningful benefit was defined as a reduction <30% from baseline to week 8 on the QIDS-C.
CAST Self-Report.
The 16 items of the CAST assess symptoms across five domains in which each individual item is rated on a 5-point Likert scale (from 1, “strongly disagree,” to 5, “strongly agree”): anxiety (three items), irritability (five items categorized as the CAST irritability domain subscale [CAST-IRR]), mania (four items), insomnia (two items), and panic (two items) (
20). Items included in the CAST-IRR are as follows: “I wish people would just leave me alone”; “I feel very uptight”; “I find myself saying or doing things without thinking”; “Lately everything seems to be annoying me”; and “I find people get on my nerves easily.” The factor structure of the CAST was reported initially by Trivedi et al. (
20) and validated by Jha et al. (
12) and Trombello et al. (
27). The CAST-IRR has been shown to have significant correlations with the Impulsivity Rating Scale (r=0.39), the Beck Anxiety Inventory (r=0.42), and the irritability item of the Clinician-Administered Rating Scale for Mania (r=0.30) (
20) as well as small to moderate correlations with comorbid psychiatric disorders (Spearman’s correlation coefficient range, 0.07–0.29) as measured by a self-report psychiatric diagnostic screening questionnaire (
12). In the present study, the Pearson correlation coefficient between the QIDS-C and the CAST-IRR was 0.35 (p<0.001) at baseline.
Statistical Analyses
The analytic sample for changes in the CAST-IRR included all CO-MED trial participants with CAST-IRR data available at baseline (N=664). Of these participants, those who had QIDS-C and CAST-IRR data at week 4 and QIDS-C data at week 8 were included in the analytic sample for prediction of acute-phase treatment outcomes (N=431). The analytic sample for replication of these predictions in SAMS included participants with QIDS-C and CAST-IRR data at baseline and week 4 and QIDS-C data at week 8 (N=163). In the CO-MED trial, repeated-measures mixed-model analyses tested baseline-to-week-4 changes in CAST-IRR score, with the visit as the within-subject variable and all other variables as between-subject variables, before and after controlling for QIDS-C score at each visit. Also in the CO-MED trial, two separate logistic regression analyses predicted remission and no meaningful benefit at week 8, with baseline QIDS-C score, baseline CAST-IRR score, percent change in QIDS-C score (100×[baseline QIDS-C score − week-4 QIDS-C score]/baseline QIDS-C score), and percent change in CAST-IRR score (100×[baseline CAST-IRR score − week-4 CAST-IRR score]/baseline CAST-IRR score) as predictor variables using the following equation:
where
p is the probability of the outcome variable (remission and no meaningful benefit at week 8), and
bi is the regression parameter for the
ith predictor. Model performance in predicting remission and no meaningful benefit after adding CAST-IRR variables were assessed using net reclassification improvement analyses (
28). The logistic regression analyses described above were repeated with sex and treatment arms (SSRI monotherapy, bupropion-SSRI, and venlafaxine-mirtazapine) separately to test whether sex and treatment significantly affected the outcomes. A receiver operating characteristic curve was plotted to obtain the area under the curve (AUC), and calibration plots (
29) were used to evaluate the agreement between predicted probabilities and observed outcomes. To generate calibration plots, the data were divided into 10 groups, the number of samples with true results equal to class was determined, and the event rate was determined for each bin. These event rates were then plotted against the midpoint of each bin. Graphically, this equates to predicted probabilities and actual outcomes falling on a 45-degree line. If a model is well calibrated, a study subject with a 0.50 estimated probability of remission will be expected to be in remission 50% of the time. The model estimates from the CO-MED trial were then used to compute individual-level probabilities in SAMS. A receiver operating characteristics plot was used to compare these predictions in SAMS with those in the CO-MED trial. An interactive calculator was developed using the Shiny package in R (
https://shiny.rstudio.com).
The threshold for statistical significance was set at a p value of 0.05. We used SAS, version 9.3 (SAS Institute, Cary, N.C.) for all analyses except the calibration plots, which were plotted in R, version 3.4.3, with the caret package, and the interactive calculator, which was developed with Shiny.
Results
Participants in both the CO-MED trial and SAMS were predominantly female (67.1% and 69.9%), Caucasian (69.2% and 71.2%), and non-Hispanic (84.2% and 90.2%). Participants in these two studies had similar baseline clinical and sociodemographic characteristics, with the exception of higher rates of chronic depression in the CO-MED trial (N=235/431, 54.5%) compared with SAMS (N=37/163, 22.7%) (
Table 1). Of the CO-MED trial participants with baseline and week-4 CAST-IRR and QIDS-C scores plus week-8 QIDS-C scores (N=431), 149 (34.6%) attained remission and 93 (21.6%) showed no meaningful benefit at week 8. Of the participants in SAMS with comparable data (N=163), 81 (49.7%) attained remission and 33 (20.3%) showed no meaningful benefit at week 8. Of the total sample in the CO-MED trial (N=665), individuals who were excluded from prediction of acute-phase outcomes (N=234) were younger, had greater depressive symptom severity at baseline, were more likely to be unemployed, were more likely to be African American, and reported less than a high school level of education (for further details, see Table S1 in the
online supplement). Similarly, in SAMS, individuals who were excluded (N=103) were younger (see Table S1 in the
online supplement).
The mean QIDS-C and CAST-IRR scores at week 4 were 9.2 (SD=4.3) and 12.6 (SD=4.6), respectively, in the CO-MED trial (N=431) and 8.6 (SD=4.4) and 12.2 (SD=4.5), respectively, in SAMS (N=163). The mean baseline-to-week-4 reduction in QIDS-C and CAST-IRR scores was 40.2% (SD=26.5) and 25.1% (SD=25.7), respectively, in the CO-MED trial and 40.8% (SD=29.6) and 21.4% (SD=29.2), respectively, in SAMS.
Change in Irritability From Baseline to Week 4
In the CO-MED trial, there was a significant baseline-to-week-4 reduction in CAST-IRR scores (F=271.80, df=3, 1663, p<0.0001; effect size=1.06) (for further details, see Figure S1 in the online supplement. This reduction in CAST-IRR scores remained significant even after controlling for QIDS-C score at each visit (F=26.17, df=3, 1661, p<0.0001; adjusted effect size=0.36). The estimated reduction in CAST-IRR scores from baseline, independent of QIDS-C change, was as follows: −1.21 (SD=0.16; p<0.0001), −1.33 (SD=0.18; p<0.0001), and −1.55 (SD=0.20; p<0.0001) at week 1, week 2, and week 4, respectively.
Change in Irritability at Week 4 as a Predictor of Remission and No Meaningful Benefit at Week 8
In the CO-MED trial, higher baseline-to-week-4 reduction in CAST-IRR scores was independently associated with higher likelihood of attaining remission (χ
2=14.60, df=1, p=0.0001) and lower likelihood of no meaningful benefit at week 8 (χ
2=4.39, df=1, p=0.036) (
Table 2). A one-standard-deviation (25.7%) greater reduction in CAST-IRR score from baseline to week 4 independently predicted a 1.73 times higher likelihood of remission and a 0.72 times lower likelihood of no meaningful benefit. Adding irritability variables to the models significantly improved the reclassification of both remission and no meaningful benefit. The net reclassification improvement for remission and no meaningful benefit was 0.36 (95% CI=0.17, 0.56, p<0.0001) and 0.34 (95% CI=0.12, 0.57, p=0.004), respectively. With the inclusion of irritability variables in the remission model, 13% of remitters were correctly reclassified, and 23% of nonremitters were correctly reclassified. Similarly, with the inclusion of irritability variables in the no meaningful benefit model, 20% of participants with no meaningful benefit were correctly reclassified, whereas 14% with meaningful benefit were correctly reclassified. Sex did not significantly predict either remission (χ
2=1.85, df=1, p=0.17) or no meaningful benefit (χ
2=0.96, df=1, p=0.33). Similarly, treatment arm did not significantly predict either remission (χ
2=1.27, df=2, p=0.53) or no meaningful benefit (χ
2=2.10, df=2, p=0.35).
Changes in Irritability and Depressive Symptom Severity as Predictors of Remission and No Meaningful Benefit at an Individual Level
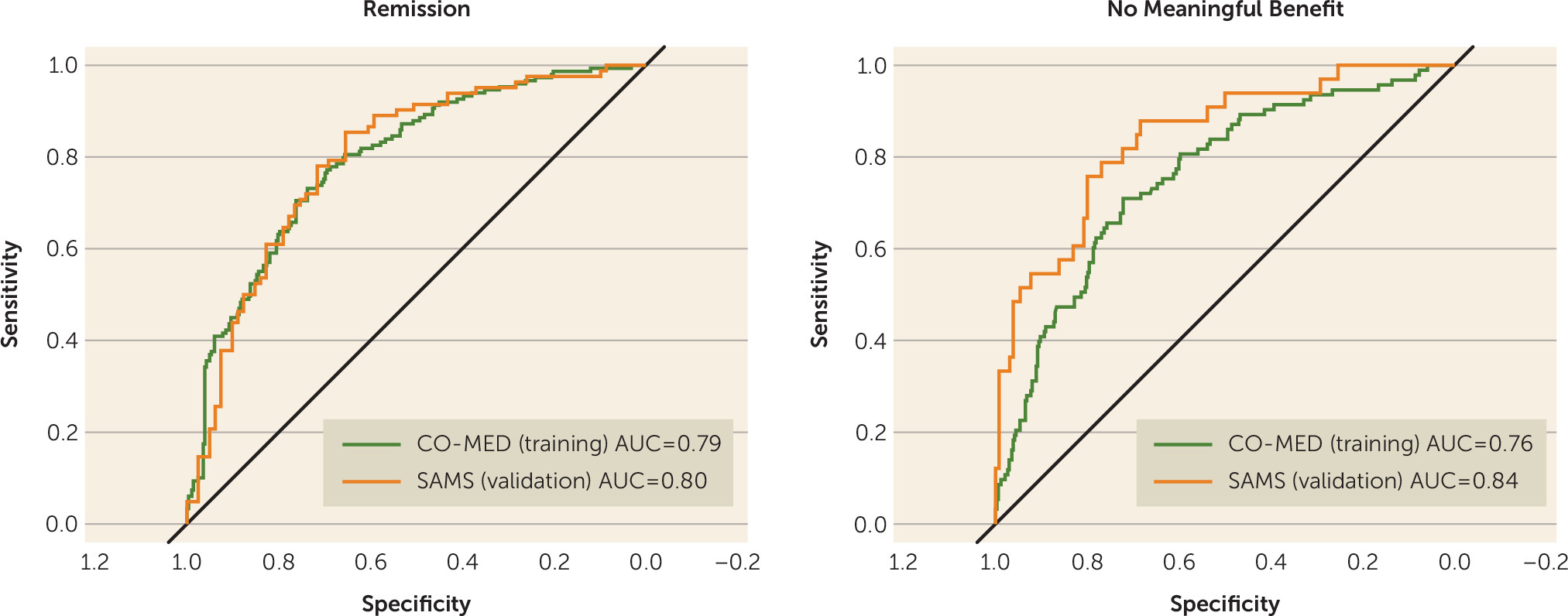
In the CO-MED trial, the model containing baseline QIDS-C and CAST-IRR scores and baseline-to-week-4 changes in QIDS-C and CAST-IRR scores had AUC values of 0.79 for remission and 0.76 for no meaningful benefit at week 8 (
Figure 1). The individual-level probabilities of remission (p/(1–p)=e
[0.6554–0.1511*(baseline QIDS-C) –0.0520*(baseline CAST-IRR) + 0.0301*(percent change in QIDS-C) + 0.0213*(percent change in CAST-IRR)]) and no meaningful benefit (p/(1–p)=e
[–1.6436 –0.00942*(baseline QIDS-C) + 0.1044*(baseline CAST-IRR) –0.0312*(percent change in QIDS-C) –0.0130*(percent change in CAST-IRR)]) were calculated. Calibration plots (see Figure S2 in the
online supplement) showed that the predicted probabilities were well calibrated, aside from the tails.
Replication of These Predictions in an Unrelated Sample of Outpatients With Major Depression
In SAMS, individual-level probabilities of remission and no meaningful benefit were obtained by using intercept and beta estimates from the CO-MED trial. In SAMS, the AUC values of remission and no meaningful benefit at week 8 were 0.80 and 0.84, respectively (
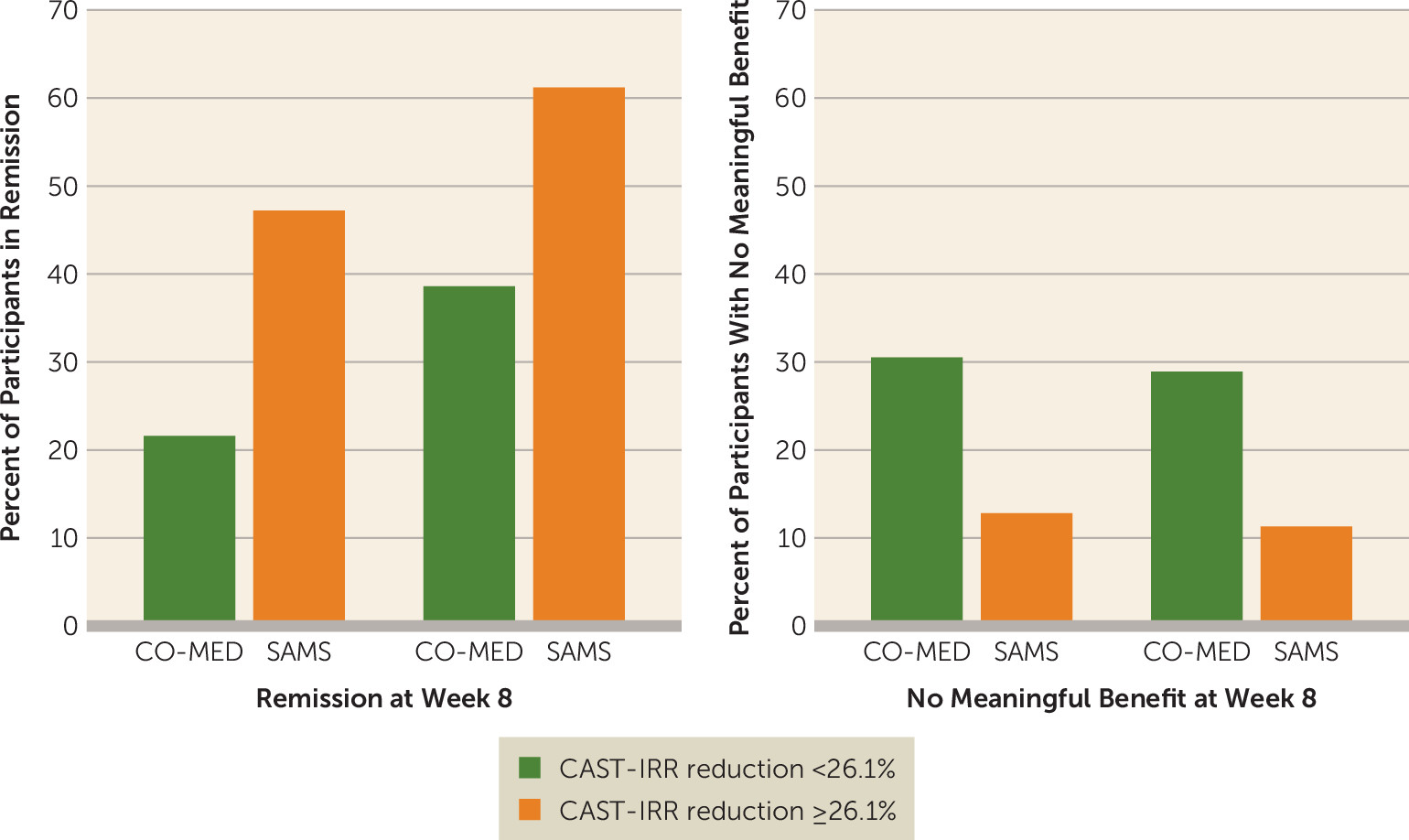
Figure 1). Using median split (the median baseline-to-week-4 CAST-IRR reduction in the CO-MED trial was 26.1%), participants were grouped by those with baseline-to-week-4 reductions ≥26.1% and <26.1% in CAST-IRR scores in order to visualize the differences in acute-phase treatment outcomes in both the CO-MED trial and SAMS (
Figure 2).
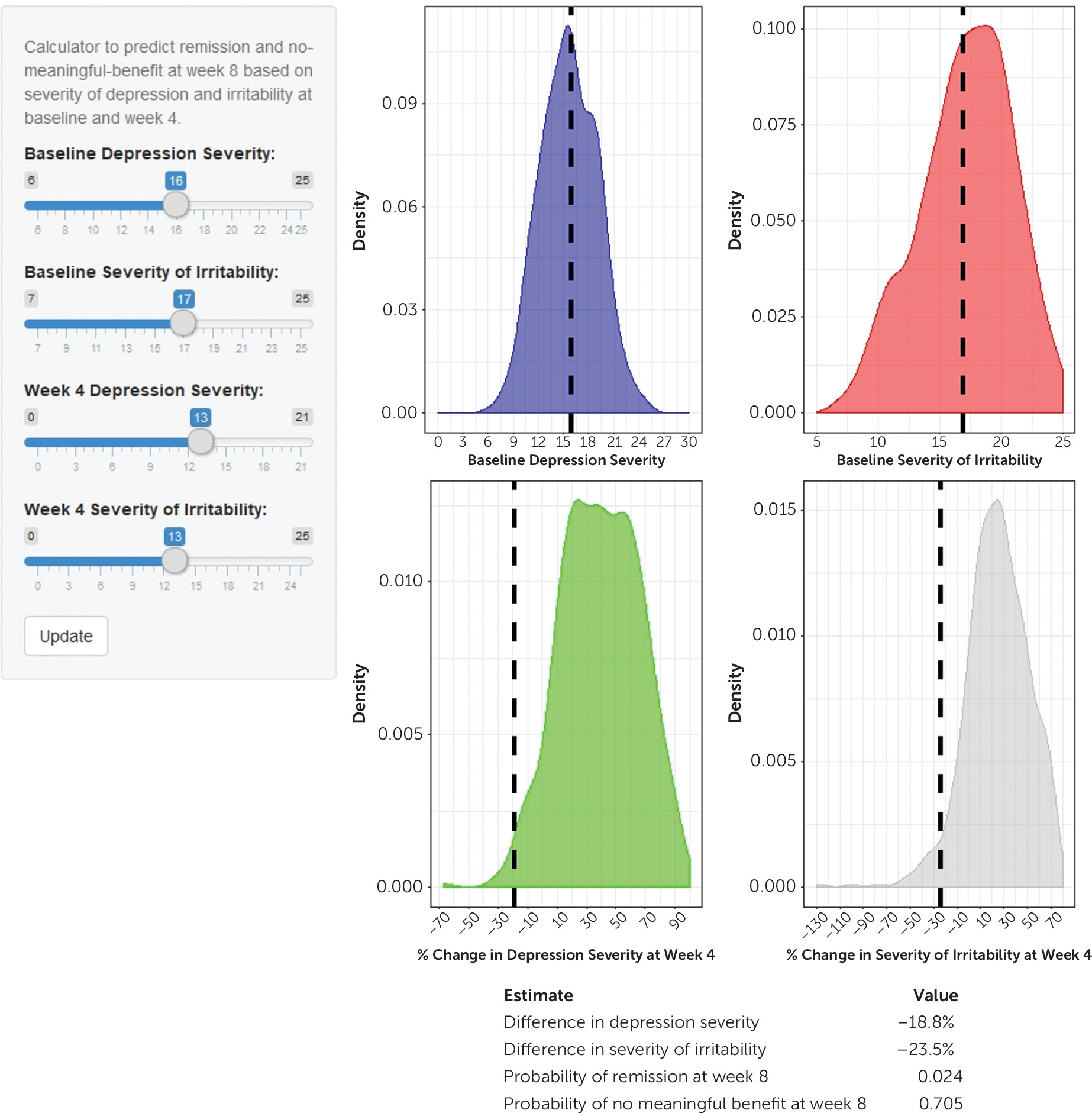
To allow estimation of individual-level probabilities, the intercepts and beta estimates from the remission and no meaningful benefit models in the CO-MED trial were incorporated in an interactive web-based calculator that could be deployed on a server for universal use. Users were able to specify the QIDS-C and CAST-IRR values at baseline and week 4, view where these individual values lay according to the distributions in the CO-MED trial, and obtain estimated probabilities of remission and no meaningful benefit at week 8 (
Figure 3).
Discussion
Irritability improved early with antidepressant treatment and predicted acute-phase treatment outcomes (remission and no meaningful benefit) independently in a large, ecologically valid sample of treatment-seeking outpatients with major depression. Furthermore, baseline-to-week-4 changes in irritability and depressive symptom severity were combined to estimate individual-level outcomes with high accuracy and were replicated in an unrelated sample. Improvement in irritability, seen as early as week 1, was not completely accounted for by reduction in depressive symptoms. Greater baseline-to-week-4 reduction in irritability was associated with higher likelihood of remission and lower likelihood of no meaningful benefit, even after controlling for baseline-to-week-4 change in depressive symptom severity and baseline levels of depressive symptom severity and irritability.
Improvement in irritability in this study is consistent with that in previous reports of reduced anger or hostility with antidepressant treatment (
13,
14). These findings also add to previous findings that improvement with antidepressant treatment extends beyond changes in core depressive symptoms (
4,
6,
7,
30). These studies, taken together, highlight the limitations of the current criteria for major depressive disorder and argue for expansion of assessments beyond the nine core diagnostic assessments. Higher likelihood of remission with greater reduction in irritability is consistent with a previous report of higher likelihood of remission with early (by week 2) improvement in anger or hostility (
14). The findings that participants excluded from the prediction models were more likely to be younger, African American, and unemployed and to have greater symptom severity at treatment initiation and less than a high school level of education are consistent with previous reports of attrition from care in the Sequenced Treatment Alternatives to Relieve Depression study (
31,
32).
A clinical implication of these findings is that irritability should be assessed during the course of antidepressant treatment. The five-item self-report measure of irritability can be implemented without significantly burdening patients and providers. Clinicians can easily combine early changes in irritability and depressive symptoms to estimate probabilities of remission and no meaningful benefit with the easy-to-use interactive calculator. The outcomes were chosen by design to be clinically actionable (for high likelihood of remission, treatment should be continued; for high likelihood of no meaningful benefit, treatment should be modified). In patients with persistent irritability at week 4 and a high likelihood of no meaningful benefit, clinicians may consider treatment strategies such as augmentation with brexpiprazole (
15).
A major strength of this study is the testing and replication of predictive models in two unrelated samples. The large sample size and the recruitment of treatment-seeking outpatients from community practices, with broad inclusion and minimal exclusion criteria, increase the generalizability of the findings.
There are several limitations to the secondary analysis. The models for remission and no meaningful benefit were tested and replicated in participants for whom a complete data set (baseline, week 4, and week 8) was available, and thus these may not generalize to individuals who dropped out of care early. As a result of the nonrandom pattern of differential attrition, the use of methods to account for missing data (such as multiple imputation) has known pitfalls (
33), and thus use of these methods was not considered appropriate. The number of predictor variables was limited to four, because the objective of this study was to demonstrate the clinical utility of adding irritability to current practices of measuring depressive symptom severity. Inclusion of other clinical and biological markers may further improve the predictive model. However, it is noteworthy that the strength of the predictive ability of the remission and no-meaningful-benefit models in SAMS (AUC values of 0.80 and 0.84, respectively) is comparable to the AUC values reported in other studies, such as the prediction of development of psychosis (AUC=0.79) in patients receiving secondary mental health care (
34) and development of bipolar spectrum disorder (AUC=0.76) in at-risk youths (
35). By design, all participants in the CO-MED trial and SAMS received a serotonergic antidepressant. Hence, these findings may not extend to individuals treated with nonserotonergic antidepressants, such as bupropion monotherapy. The individual-level calculator is restricted by the choice of the QIDS-C and the CAST-IRR as measures of depression severity and irritability. Further studies are needed to test the validity of this calculator with other measures of depression severity and to evaluate whether the measurement-based care paradigm that incorporates assessments of irritability along with depression severity results in improved treatment outcomes. The model is well calibrated with probabilities ≤0.6 in both models, and thus any estimates outside this range should come with the understanding that they may not produce outcomes at the anticipated rate. This low calibration may be a result of the fact that only about 20% of the CO-MED trial participants in the no-meaningful-benefit model were assigned probabilities above 0.6, and only 5% of the CO-MED trial participants in the remission model were assigned probabilities above 0.5. More information (including additional predictors and larger samples) is needed to better gauge calibration accuracy in these groups. Calibration plots have a major limitation—they represent all binned data as if there were equal amounts of data in each bin, which often is not the case.
In conclusion, irritability improves early with antidepressant treatment independently of depressive symptom severity. This early improvement is independently associated with higher likelihood of remission and lower likelihood of no meaningful benefit. The combinations of baseline and early changes (up to week 4) in irritability and depressive symptom severity can estimate individual-level probabilities of remission and no meaningful benefit. These findings support inclusion of assessments of irritability in measurement-based-care approaches for the treatment of patients with major depression.
Acknowledgments
The authors thank the clinical staff at each clinical site for their assistance, all of the study participants, and Eric Nestler, M.D., Ph.D., and Carol A. Tamminga, M.D., for administrative support.