The accurate diagnosis of bipolar disorder in young people is a challenge, in part because acute symptoms are often nonspecific and overlap with other disorders, and the illness course appears to be influenced by both developmental and clinical stage (
1,
2). Delayed recognition and treatment of bipolar disorder contributes to the associated substantial burden and increased mortality (
3,
4). Evidence from cognitive, imaging, and biological studies supports the observation that persistent functional deficits and structural and functional anomalies generally occur after the onset of full-blown bipolar disorder, in contrast to the developmental trajectory of schizophrenia, which is characterized by significant functional and structural decline prior to the first psychotic episode (
5–
7). Collectively, these findings underscore the importance of improving accurate early detection of emerging bipolar disorder, with the overall aim of preventing illness progression (
8,
9).
Bipolar disorder runs in families, as reflected by high heritability estimates (
10,
11). Children of parents with bipolar disorder are therefore an identifiable high-risk group that can inform the effort to characterize the emergent illness course. Since our first publication from the Canadian high-risk offspring cohort study in 1998 (
12), several longitudinal high-risk offspring studies have been launched around the world (
13–
16). These studies have distinctive differences in methodology, including the approach to recruiting and assessing parents, contributing to differences in rates of comorbidity in affected parents and psychiatric illness in the nonproband parent (for a review, see reference
17). Yet all studies have observed significantly higher rates of a broad spectrum of lifetime psychiatric disorders in high-risk offspring, despite the specific familial risk for bipolar disorder. Several studies have highlighted the prominence of depressive disorders in the early course of illness (
13,
16,
18), and findings from both the Amish and the Pittsburgh studies indicate that internalizing symptoms, mood lability, and, in the latter case, proximal hypomanic symptoms may be predictors of bipolar disorder in children at familial risk (
16,
19).
Over the past two decades, we have continued to prospectively study the children of bipolar parents subtyped by parental response or nonresponse to long-term lithium treatment (
12,
18,
20). Specific parental illness characteristics and subtypes can be useful in risk prediction and in identifying a prototypical illness trajectory—a prerequisite for mapping biomarkers and identifying specific intervention targets (
21,
22). The lithium-responsive phenotype identifies a more homogeneous bipolar disorder subtype that is consistent with classical manic-depressive illness and is characterized by a highly recurrent course prior to lithium stabilization, prominence of depressive episodes, classical euphoric manias, good quality of spontaneous remission, low comorbidity, and specific genetic, neural, and neurophysiologic associations (
6,
23–
29). We previously provided a description of the development of bipolar disorder in high-risk offspring subtyped by parental response to lithium prophylaxis and estimated a preliminary model using syndrome-level data (
20).
Here, 20 years after our first publication on this topic (
12), we present a comprehensive analysis of the antecedent and emergent clinical course of bipolar disorder over a longer period of observation, using a larger number of high-risk offspring and comparing subgroups based on parental response or nonresponse to lithium prophylaxis. This analysis considers the association between parent and offspring course as well as both clinically significant symptom-level data and syndrome-level data. Our objectives were to compare rates of lifetime psychopathology in high-risk offspring and control subjects and between high-risk offspring subgroups; to describe the early course of emergent bipolar disorder and compare it with the parents’ course; to estimate the strength of associations between antecedent symptom- and syndrome-level psychopathology and mood disorder; and to model the trajectory of emergent bipolar disorder using symptom- and syndrome-level data.
Methods
Study Families
The Flourish Canadian high-risk study is a dynamic, open prospective cohort study that started in 1996 (
12,
20). The study was approved by the Ottawa Independent Research Ethics Board and the Queen’s University Health Sciences Research Ethics Board. Families were identified through mood disorder subspecialty clinical programs in Ottawa and the Maritimes (
12,
24). Bipolar parents were participating in clinical, genetic, and neurobiological research and were treated systematically and prospectively within the clinical programs. A lifetime diagnosis of bipolar I or II disorder was based on semistructured research interviews using the Schedule for Affective Disorders and Schizophrenia–Lifetime version (SADS-L), conducted by a psychiatrist and confirmed in blind consensus review using all available clinical and research information by at least two independent research psychiatrists. We subsequently expanded the sample of study families, using the same methods to recruit adult siblings of the original bipolar probands who were themselves affected with either bipolar disorder or recurrent major depressive disorder, adding an additional 77 high-risk offspring.
Response to prophylactic lithium treatment in affected bipolar adults was defined as having no new recurrences over a minimum observation period of 3 years while on therapeutic blood levels of lithium after a highly recurrent pretreatment course (
24). In affected adults not treated by us, lithium response or nonresponse was estimated using a validated scale that weighs completeness of lithium response against potential confounding factors, such as shorter duration of treatment, use of additional medications, and adherence (
21). For inclusion in this study, the other biological parent was confirmed to have no lifetime major psychiatric disorder (i.e., schizophrenia, bipolar disorder, or recurrent major depressive disorder) at the time of recruitment, based on SADS-L interview or family history provided by the proband.
Comparison families were recruited from schools in Ottawa reflecting socioeconomic backgrounds and geographic area similar to those of the high-risk families. As described elsewhere (
20), families with children in grades 6–12 were mailed a demographic screening questionnaire by the school. At least one parent from interested families was assessed by a research psychiatrist using the SADS-L interview (
18,
20). For parents not directly assessed, information from the screening questionnaire and the interviewed parent was used to determine eligibility. All clinical diagnoses in parents were confirmed by blind consensus reviews involving two research psychiatrists. For inclusion in the study, control parents were confirmed to have no lifetime major psychiatric disorder at the time of recruitment.
Study Offspring
Eligible offspring were from identified high-risk and control families, were in the age range of 5–25 years at baseline, and were able to comply with the study protocol (i.e., not suffering from a serious neurological disorder or intellectual disability). All offspring completed research assessments at baseline and then, on average, annually thereafter. Baseline and subsequent assessments included a semistructured interview following the SADS-L format or the Kiddie Schedule for Affective Disorders and Schizophrenia–Present and Lifetime version (K-SADS-PL), administered by a psychiatrist blind to study group, along with validated self-report and clinician measures of dimensional symptoms, global functioning, and other exposures described below. For offspring who could not attend a follow-up assessment in person, we used video call interviews.
All diagnoses were based on DSM-IV criteria, and clinically significant subthreshold symptoms were based on operationalized definitions (see the online supplement) using all available research and clinical information (i.e., prior consultation and psychoeducational evaluations) and using the best-estimate procedure in blind consensus reviews that included at least one additional research psychiatrist and a clinician with graduate-level training. While this is a naturalistic observational study, in the event of illness onset, offspring were offered a clinical consultation and recommendations based on published treatment guidelines were sent to a responsible physician. In these instances, blinding was broken for a research psychiatrist to complete the consultation; however, the blind was maintained for consensus reviews.
Measures
At baseline, the Hollingshead socioeconomic scale (
30) was completed for all parents. Childhood abuse was measured in offspring age 13 and older with the Childhood Experience of Care and Abuse Questionnaire (
31). Global functioning was assessed by a clinician at each visit, using the Global Assessment of Functioning Scale (GAF) (
32).
Statistical Analysis
Cumulative incidence was calculated to estimate the expected proportion of subjects who would meet the criteria for lifetime diagnoses by the last assessment in the study; estimates were made nonparametrically, accounting for variable age at first and last observations. Cox proportional hazard models were used to test for differences in hazard ratios between groups. Firth’s penalized method and Breslow’s method for handling ties were used when necessary to account for low rates of psychopathology among control subjects. Differences in median age at onset of psychopathology were estimated using accelerated failure time models. To estimate the hazard of bipolar disorder, given prior clinical symptoms and other diagnoses, Cox proportional hazard models with time-varying covariates, adjusted for sex and sibling correlation, were calculated, producing hazard ratios and 95% confidence intervals. To estimate associations between parent clinical course (i.e., age at onset, nature of clinical course) and offspring clinical course, generalized estimating equations and generalized linear mixed-effects models, adjusted for sex and familial clustering, were used.
To test our proposed model of emergent bipolar disorder, three versions of a parametric multistate model were fitted: an unrestricted model that allows subjects to transition between any of the “stages” of illness; a forward model that only allows subjects to transition into higher numbered stages; and a progressive model that only allows transitions to the next stage in the sequence. To accommodate observations that violate the proposed model structures, latent misclassification rates were included in all the proposed models (
33). To assess how subthreshold symptoms influenced the progression in the model, a semiparametric Cox proportional hazard–type multistate model was fitted, with time until transition to the next stage as the outcome. Each transition was represented by a separate proportional hazard model, with onset of subthreshold symptoms as predictors in the models along with sex (see Table S3 in the
online supplement). We maintained the 0.05 alpha level with no adjustments for multiple comparisons, given the observational nature of the study and in order to limit the probability of unnecessary type II errors. All statistical analyses were conducted using SAS, version 9.4, except for the parametric multistate models, which were fitted using the msm package in R, version 3.3.2.
Results
Sample Description
This analysis included 116 high-risk and 55 control families contributing a total of 279 high-risk offspring (117 from lithium-responsive parents and 162 from lithium-nonresponsive parents) and 87 control subjects, reflecting an additional 50 high-risk offspring since our last report on this cohort (
20). Up until last assessment, 12 high-risk offspring could not be contacted or dropped out (seven from lithium-responsive and five from lithium-nonresponsive parents), representing a loss to follow-up of <5% during the study period. The median time between study assessments was 1.58 years. As shown in
Table 1, the ages at baseline and last assessment for high-risk offspring were 16.48 years (SD=6.26) and 23.61 years (SD=8.14), respectively. The number of follow-up study years varied, given the open, dynamic cohort design, and ranged from 1 to 21 years for high-risk offspring (mean=7.72, SD=5.28), representing up to an additional 5 years of observation since our last report (
20).
Demographic and illness course data for the parents are summarized in Table S1 in the online supplement. Parents with bipolar disorder that responded to lithium prophylaxis had less lifetime comorbidity compared with those whose illness was nonresponsive to lithium (p=0.036). Specifically, lithium-responsive parents had lower rates of substance use disorders (p=0.022) and marginally lower rates of anxiety disorders (p=0.074) and lifetime psychotic symptoms in mood episodes (p=0.017) compared with lithium-nonresponsive parents. Lithium-responsive parents also had a higher rate of a completely remitting illness course compared with lithium-nonresponsive parents (<0.001).
As shown in
Table 1, most high-risk offspring and control subjects came from middle- to upper-middle-class families with relatively high rates of family intactness over the first decade of life (77.8% and 85.1%, respectively [p=0.15]) and comparably low rates of physical or sexual abuse in childhood (17.4% and 13.6%, respectively [p=0.65]). High-risk offspring had lower GAF scores at baseline (p<0.001) and at the most recent assessment (p<0.001) compared with control subjects. Post hoc analysis revealed that the difference in GAF scores at the last observation was accounted for by a decline in GAF scores among the high-risk offspring of lithium-nonresponsive parents.
Lifetime Psychopathology
Table 2 summarizes the cumulative incidence of hierarchically defined DSM-IV lifetime psychiatric disorders and subthreshold symptoms in the high-risk offspring and control groups. Bipolar and psychotic disorders were observed only in the high-risk offspring and not in the control group. There were no differences in the rate of bipolar disorder between high-risk offspring subgroups; however, psychotic disorders manifested almost exclusively among the offspring of lithium-nonresponsive parents (20.22% compared with 1.02% [p=0.025]). Depressive disorders were more frequent than bipolar disorder across study groups. Major depressive disorder and sleep disorders were almost exclusively observed in the high-risk offspring (32.93% and 4.88% [p=0.003], and 23.19% and 0% [p=0.013], respectively, in the high-risk and control groups). There were no significant differences in the cumulative incidence of lifetime anxiety disorders, attention deficit hyperactivity disorder (ADHD), learning disorders, adjustment disorders, and substance use disorders between high-risk offspring and control subjects by last observation. Anxiety and adjustment disorders were higher among the offspring of lithium-nonresponsive than lithium-responsive parents, estimated at 45.37% and 27.82% (p=0.035), respectively, and 60.12% and 28.34% (p=0.099), respectively. As shown in Figure S1A and S1B in the
online supplement, anxiety and substance use disorders tended to manifest earlier in high-risk offspring, but with longer observation time the risk increased to comparable levels among control subjects. The cumulative incidences of subthreshold depressive and hypomanic symptoms by age at last observation were higher among high-risk offspring compared with control subjects, estimated at 16.18% and 1.49% (p=0.024), respectively, and 22.01% and 1.69% (p=0.017), respectively. The cumulative incidence of subthreshold symptoms was comparable between the high-risk offspring subgroups (
Table 2).
Table 3 compares the median age at onset of hierarchical mood and non-mood diagnoses and subthreshold symptoms between study groups. Onset of bipolar disorder tended to be in late adolescence or early adulthood (median=20.73 years; range=12.37–30.25). There was no instance in which the diagnostic criteria for bipolar disorder were met in a child before age 12 (as defined by age at first meeting full DSM criteria). Depressive disorders and major depressive disorders occurred at a significantly lower median age in high-risk offspring compared with control subjects (15.80 years and 18.45 years [p=0.037], respectively, and 17.54 years and 20.33 years [p<0.001], respectively). There were no differences in median age at onset of mood disorders between high-risk offspring subgroups. The median age at onset of non-mood psychopathology tended to be lower in high-risk offspring compared with control subjects; median age at onset of substance use disorders was lower in the offspring of lithium nonresponders compared with the offspring of lithium responders, although the difference fell short of statistical significance (16.15 and 16.85 years, respectively [p=0.059]). The median age at onset of subthreshold mood symptoms in high-risk offspring tended to be lower than in control subjects. There were no differences in age at onset of these symptoms between high-risk subgroups.
Early Course of Bipolar Disorder
In high-risk offspring who met DSM-IV criteria for a bipolar spectrum disorder (bipolar I and II disorders, bipolar disorder not otherwise specified, and schizoaffective bipolar disorder) by last observation, we had sufficient data to examine the polarity of the first five mood episodes. More than 85% of index mood episodes were depressive, and the depressive polarity remained prominent over the first five bipolar mood episodes (see Table S2 in the online supplement). A generalized linear mixed-effects model (see Figure S2A in the online supplement) provided evidence that polarity of the first five mood episodes of bipolar disorder was associated with high-risk subgroup (p=0.04). Specifically, offspring of lithium-nonresponsive parents had lower odds of the mood episode being depressive over subsequent episodes (p=0.030), but this was not true for offspring of lithium-responsive parents (p=0.41). The risk of psychotic features in mood episodes was higher among offspring of lithium-nonresponsive than among offspring of lithium-responsive parents (see Figure S2B in the online supplement). Furthermore, there was strong evidence that the clinical course of bipolar disorder differed between the high-risk subgroups. That is, the offspring of lithium-responsive parents were more likely to have a fully remitting clinical course, whereas the offspring of lithium-nonresponsive parents were more likely to have a remitting course with residual symptoms or a chronic, fluctuating course (p<0.001).
There was evidence that earlier age at onset in the bipolar parent was associated with earlier age at mood disorder onset in the offspring (p=0.014). Specifically, for every 1-year decrease in the parents’ age at onset, there was a 0.12-year decrement in mean age at onset in the offspring. Furthermore, there was evidence that clinical course of the parent was associated with clinical course of the offspring (p<0.001). It was estimated that parents with a fully remitting bipolar disorder were more likely to have children with a fully remitting mood disorder compared with parental bipolar disorder with residual symptoms (p=0.009) or a chronic, fluctuating course (p=0.019). However, the presence of lifetime psychotic features in parents was not statistically associated with the presence of psychotic features in the offspring (p=0.65).
Estimating the Risk of Mood Disorder With Antecedent Psychopathology
The adjusted hazard of developing a mood disorder was greater among high-risk offspring with an anxiety (hazard ratio=1.84, p=0.002) or sleep disorder (hazard ratio=1.63, p=0.044) compared with those without (
Table 4). In addition, antecedent subthreshold depressive and hypomanic symptoms increased the hazard of mood disorder by 2.67 (p<0.001) and 2.34 (p=0.045) times, respectively. While subthreshold depressive symptoms were significantly associated with the hazard of any mood disorder among both high-risk males and females, the magnitude of risk was much higher for males (hazard ratio=4.76, p<0.001) compared with females (hazard ratio=1.99, p=0.020). Furthermore, the hazard of any mood disorder was increased by 4.52 times for males (p=0.019) with prior subthreshold activation, which was not statistically significant in females.
Estimating the Risk of Mood Disorder Based on Parental Illness Variables
In a Cox proportional hazard model adjusting for familial clustering and sex, there was no evidence that parent age at onset (p=0.35), parent clinical course (p=0.51), or parent lifetime psychotic features (p=0.52) increased the hazard of bipolar disorder in high-risk offspring. However, there was marginal evidence (p=0.053) that younger parent age at onset increased the hazard of mood disorders, including bipolar disorder, in high-risk offspring (hazard ratio=1.02, 95% CI=1.00, 1.04).
Model of Clinical Trajectory of Emergent Bipolar Disorder in High-Risk Offspring
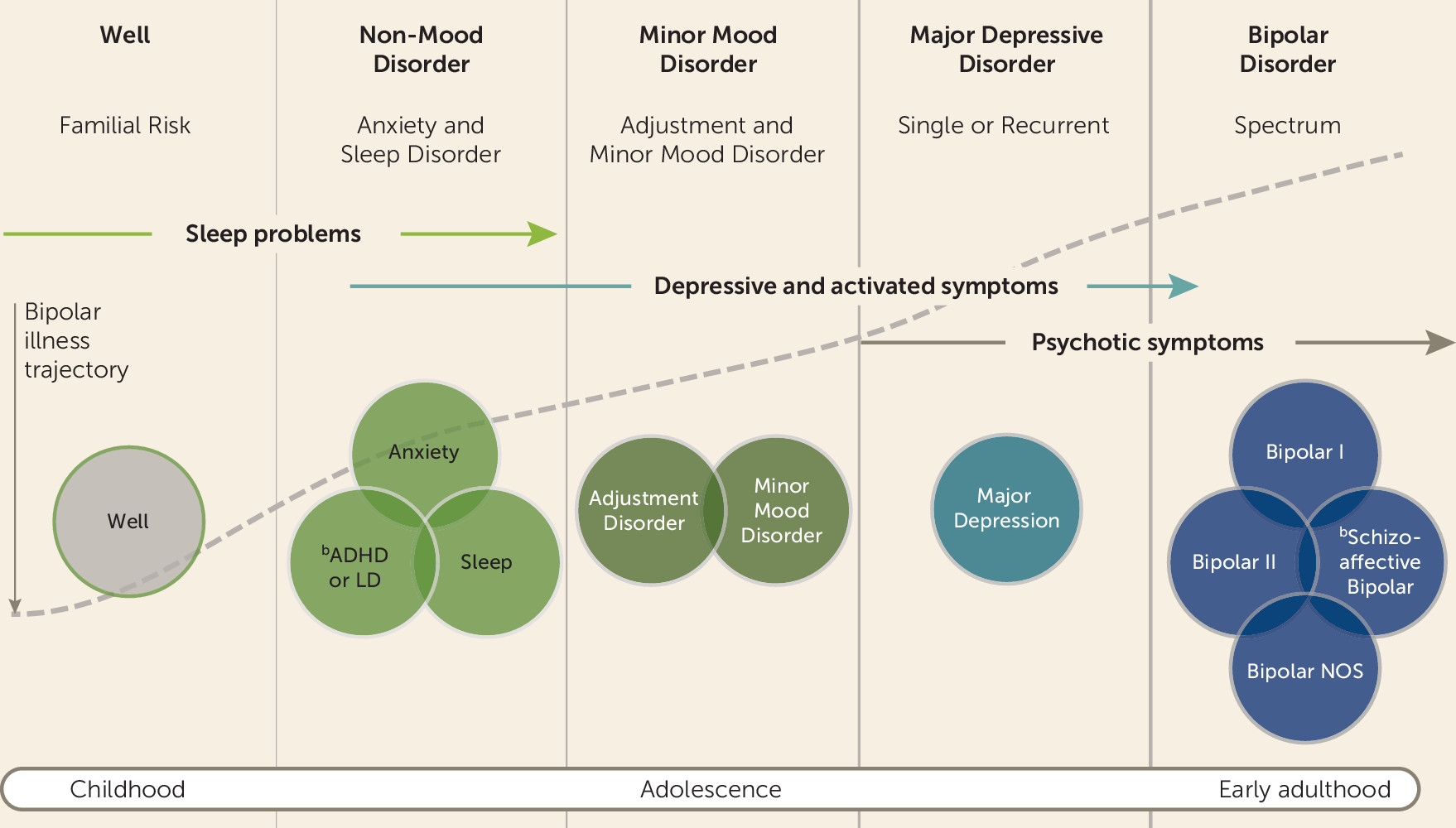
We defined each illness transition or “stage” as follows: stage 0, well but at familial risk; stage 1, non-mood disorders, including sleep disorders, anxiety disorders, ADHD, and behavioral disorders; stage 2, minor mood disorders, including adjustment disorders, depression not otherwise specified, mood disorder not otherwise specified, dysthymia, and cyclothymia; stage 3, major depressive disorder, single or recurrent; and stage 4, bipolar disorder, including bipolar disorder I and II disorders, bipolar disorder not otherwise specified, and schizoaffective bipolar disorder (
Figure 1). In this analysis, the progressive model (Akaike information criterion [AIC]=2726.82) was selected over the unrestricted model (AIC=2759.21) or the forward model (AIC=2738.96). There was evidence (see Table S3 in the
online supplement) that subthreshold sleep symptoms were associated with an increased risk of transitioning from well to non-mood disorder (p=0.036) after adjusting for other subthreshold symptoms, and that high-risk females had a higher risk of progressing from non-mood disorder to minor mood disorder (p=0.004) and from minor mood disorder to major depressive disorder (p=0.005) compared with high-risk males (see Table S3). Finally, psychotic features in mood episodes were associated with an increased risk of transitioning from major depression (stage 3) to bipolar disorder (stage 4) in the model (p=0.010).
Discussion
We report new and expanded observations about the developmental trajectory and early course of bipolar disorder in high-risk offspring, including a novel comprehensive multivariate analysis considering both symptom- and syndrome-level psychopathology. Offspring were subgrouped by parental response or nonresponse to lithium prophylaxis, allowing comparisons between offspring from parents with different bipolar subtypes. This latter point is important because bipolar disorder is highly heterogeneous and includes a number of different illness subtypes, likely each with a distinct etiology and emergent clinical course (
25,
28,
29,
34).
Lifetime Psychopathology
Bipolar disorder manifests in high-risk offspring and not in control subjects even with longer follow-up. At last observation, the cumulative incidence of bipolar disorder approached 25%, representing a small increase from our last report on this cohort, estimated at 22% (
20). Consistent with reports from a number of prospective offspring studies, age at onset of bipolar disorder was concentrated in late adolescence and emerging adulthood and ranged from ages 12 to 30 (
13,
15–
17,
35). We have not observed a single case of full-blown mania or hypomania in our offspring cohort younger than age 12, despite having a parent with a stable (assessed up to 40 years) bipolar diagnosis, and often deriving from large, multigenerational families with high illness penetrance. Similar findings have been reported by other independent high-risk offspring studies (
13,
15,
16,
35), but not all (
36). As reviewed in detail elsewhere, reported manic/mixed and hypomanic episodes in young children may reflect differences in the families under study, methods of offspring assessment, and the clinical interpretation of broad-spectrum irritability or manic-like symptoms, often in the context of ADHD (
1,
17,
37).
Also, consistent with previous observations, we found depressive disorders to be much more common than bipolar disorder in high-risk offspring, with an overall cumulative incidence of around 70%, representing an increase from our last report of 61% (
20). While the total cumulative incidence of depressive and adjustment disorders did not differ between the high-risk offspring and control groups, major depressive disorder (at least to this point in development) manifests almost exclusively in the high-risk offspring and not in the control group. Moreover, age at onset for all depressive disorders was earlier in high-risk offspring compared with control subjects, especially for major depressive disorder, with a median onset in mid to late adolescence. It is likely that with longer observation, the cumulative incidence of major depressive disorder will increase in the control group.
The cumulative incidence of both anxiety and substance use disorders in control subjects significantly increased since our last report, and at last observation it was comparable to that observed in the high-risk offspring. The cumulative incidence of childhood sleep disorders remained significantly elevated in high-risk offspring compared with control subjects. The median ages at onset of non-mood disorders tended to be earlier in high-risk offspring compared with control subjects, but only substance use disorders maintained significance after adjustments (16.7 and 19.7 years, respectively). We continue to observe relatively low and comparable rates of ADHD and behavioral disorders in both the high-risk offspring and control groups. This observation is in line with some high-risk studies (
13,
16) but differs from others (
14). As discussed elsewhere (
17), this finding in part reflects methodological differences among studies in recruitment and assessment, nonspecific psychosocial factors (intactness and socioeconomic status of families), comorbidity in the bipolar parent, and prevalence and nature of psychiatric illness in the other biological parent.
There were few differences between the high-risk offspring subgroups in cumulative risk and age at onset of lifetime disorders. However, among the offspring of lithium-nonresponsive parents, there was a marginally higher incidence of adjustment disorders, perhaps signifying more vulnerability under stress, as well as a significantly higher incidence of lifetime anxiety and psychotic disorders. This is consistent with findings from genetic and neurobiological studies showing differences between lithium-responsive and lithium-nonresponsive bipolar subtypes (
38,
39). A recent genome-wide association study found an inverse relationship between genetic loading for psychotic illness risk variants and lithium response in adult patients with bipolar disorder (
40). Over this longer observation time, a decline in global functioning among the offspring of lithium nonresponders was observed, while the well or remitted offspring of lithium responders maintained a stable and high level of global functioning.
Early Course of Bipolar Disorder
Across both high-risk offspring subgroups, bipolar disorder almost always debuted with a depressive mood episode. Furthermore, depressive episodes dominated the early bipolar course in both high-risk subgroups. This prominence of depressive episodes early in the course of bipolar disorder has also been reported in the Dutch longitudinal high-risk study (
13). In comparisons between the high-risk subgroups, the bipolar offspring of lithium responders were more likely than those of lithium nonresponders to have depressive episodes over the early course, an episodic and completely remitting illness course, and a lower risk of psychotic symptoms in mood episodes. Furthermore, there was a significant association between the nature of the parent’s course of bipolar illness and that of the affected offspring. This further supports the supposition that this subtype breeds true from parent to child.
Antecedent Syndromes and Symptoms
We found evidence that childhood anxiety disorder predicted an almost twofold increase in risk of subsequent mood disorder, consistent with our previous report (
41). Similarly, there was evidence that childhood sleep disorder predicted over a 1.6-fold increase in risk of subsequent mood disorders. Furthermore, we showed that clinically assessed subthreshold depressive and manic symptoms predicted over 2.7-fold and 2.3-fold increases, respectively, in risk of mood disorder in high-risk offspring. These findings are in line with a recent report from the Pittsburg group showing the predictive importance of self-reported or parent-reported mood and anxiety symptoms in high-risk children (
19) and with similar findings from the Amish high-risk cohort (
16). The implication is that clinically significant anxiety, mood, and sleep symptoms in children at confirmed familial risk for bipolar disorder identify an ultra-high-risk group that likely warrants closer surveillance and support. These clinical manifestations appear to be important early intervention targets worthy of systematic study. Consistent with findings from a Swiss offspring study (
15), we found evidence that earlier age at onset of parental bipolar disorder was associated with an increased risk of mood disorder in their offspring.
Model of the Developmental Trajectory of Bipolar Disorder in High-Risk Offspring
Findings from this analysis expand on earlier findings from this cohort (
20) and from a Dutch high-risk offspring study (
13) showing that bipolar disorder typically debuts with single or recurrent major depressive disorder in adolescents at familial risk. Furthermore, evidence supports predictive significance of childhood sleep and anxiety disorders as well as antecedent clinically significant mood symptoms of both polarities for subsequent mood disorder in high-risk children (
20,
41). Our novel multistate model estimating the trajectory of emerging bipolar disorder is consistent with a progressive sequence from nonspecific childhood disorders to minor mood disorders and then major depressive disorder and finally full-blown bipolar disorder in emerging adulthood. While individuals may not manifest all illness stages in the trajectory, when they join the model they tend to transition from that point forward in sequence. We found evidence that sleep symptoms increase the likelihood of transition from well to childhood non-mood disorders, and psychotic features in depressive episodes increased the likelihood of transition to full-blown bipolar disorder, consistent with previous observations in clinical samples (
42). However, we also found evidence of significant heterogeneity in the emergent course of bipolar disorder, as indicated by the variation in age at onset of psychopathology at both the symptom and syndrome levels and in the hazard ratios and confidence intervals within and between high-risk subgroups. Furthermore, the clinical course appears to breed true from bipolar parent to affected child, with the classical lithium-responsive illness characterized by an episodic, recurrent course with good quality of remission, a prominence of depressive episodes early in the course, and stable rather than declining global functioning.
Limitations
Our study has several limitations. First, it was specifically designed to describe the developmental trajectory of bipolar disorder in two distinct high-risk offspring subgroups that would inform future studies of associated biomarkers and genetically sensitive pathways to illness onset. We therefore limited inclusion to families with only one affected parent with well-characterized bipolar disorder to minimize the confounding effects of assortative mating. Consequently, our findings may not generalize to heterogeneous clinical, help-seeking, or general-population samples. Second, given our dynamic design, some episodes occurred prior to baseline and were identified retrospectively. However, these episodes were confirmed using a best-estimate diagnostic approach, verified with prospectively captured clinical reports, limiting recall bias. Although the GAF is a complex measure that limits the assessment of the contribution of specific domains (i.e., clinical and psychosocial), we used it as a global measure of stability of functioning repeatedly over time. Finally, while this is a naturalistic study, treatment could have influenced outcomes. However, we estimated, on the basis of systematic documentation at study visits, that medication likely had a minimal impact on the natural early course of bipolar disorder (analyzed over the first five diagnosable mood episodes). That is, mood stabilizers, continued after the remission of the first acute mood episode, were used in around 10% of participants, and after the remission of the second and third episodes, in an estimated 30% of participants.
Implications
This study has several implications. First, the study findings are in accordance with those from other independent high-risk offspring studies, and they underscore the importance of taking into account both the family history and the developmental trajectory of emerging psychopathology to improve earlier diagnostic precision in young people manifesting clinically significant symptoms and syndromes. Second, the findings emphasize the importance of antecedent anxiety and sleep disorders as well as major depression (especially with psychotic symptoms) in the emergent course of bipolar disorder in young people at confirmed familial risk—which can complicate early recognition and contribute to paradoxical or seemingly refractory response to antidepressant treatment. Third, early clinical intervention and prevention efforts should emphasize low-risk interventions addressing mood symptoms, anxiety and sleep disorders, and prevention of substance misuse. Finally, to improve our understanding of the risk processes contributing to the onset of bipolar disorder and to map biomarkers to the emergent course, there is a need to prospectively study individuals at confirmed high risk, starting from well-characterized parents who have stable and valid diagnoses, and to take into account the significant heterogeneity of bipolar disorder as it is currently defined—that is, to study more homogeneous subgroups. This study supports consistency in phenotype across generations, implicating shared underlying etiology and treatment response.
Acknowledgments
Supported by a grant from the Canadian Institutes of Health Research (PJT 152976).