The search for genetic risk factors for schizophrenia specifically and psychotic disorders more broadly has been advancing at rapid speed. In our enthusiasm for this line of research, we are at risk of minimizing the importance of environmental risk factors. The article by Lee et al. in this issue of the
Journal (
1) is thus a salutary reminder that environmental risk factors are of potential substantial significance in these disorders and may offer an opportunity to identify potential targets for primary prevention.
Lee et al. build on a long line of research that began by examining increased rates of schizophrenia in women who were pregnant during flu epidemics (
2). An important methodological advance in this field was moving from these ecological studies to those that directly documented the risk exposure of individual mothers, an approach utilized by Lee et al. in this study. The study is also part of the effort to clarify the boundaries of pathogenic exposures, in this case expanding from viral to bacterial infections.
Given the establishment, as documented in this article, of an association between a putative risk factor (prenatal bacterial infection) and an outcome (psychotic disorders in the offspring, including psychotic affective illness), the next important—and often difficult—question is the degree to which this association reflects causal effects.
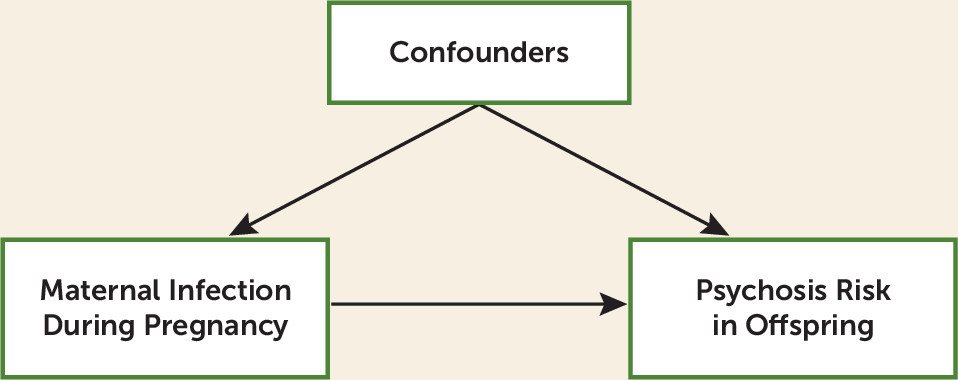
Figure 1 is a typical schematic of the problem. Is the association between maternal infection and risk for schizophrenia largely a result of a direct causal path from risk factor to illness or the result of a range of possible background confounding factors that predict risk both for maternal infection and schizophrenia in offspring?
This is an important question for two reasons. First, only causal effects can be successfully intervened upon. One quite attractive feature of this study is the potential for translating these results into primary prevention—that is, careful screening of vulnerable pregnant women for bacterial infections followed by rapid and aggressive treatment. However, any such effort can succeed only if a substantial proportion of the observed association is indeed causal. Second, another goal for these studies is to identify etiologic mechanisms that could lead to currently unimagined primary prevention approaches or even treatments. If these observations reflect causal processes, then large-scale biological studies to clarify such mechanisms would likely be a prudent investment. But it will be a fool’s quest if the original association results from confounding factors.
The authors’ primary statistical approach to examine the association between their exposure and outcome is multiple regression, which traditionally does not get high marks as a causal inference method (
3). That is because causal effects can be isolated by multiple regression only if the regression model includes good measures of all the important potential confounding variables. This is a difficult claim to meet with confidence, as it is hard to be confident that all the confounders have been identified and well measured. To their credit, the authors utilize a substantial list of covariates in their “adjusted analysis” that appear to have been carefully selected to include key potential confounders, such as the mother’s socioeconomic status and a parental history of psychiatric illness. However, confounding can arise even if key covariates are included but are measured inadequately. For example, the authors note the possible concern about parental familial/genetic risk for psychosis predisposing to infections and also acknowledge that their measure of parental psychopathology is far from ideal. It is worth noting that the aggregate effect of all their chosen covariates on the risk factor–outcome association is rather modest, which is somewhat reassuring.
As is often the case in psychiatric epidemiology, the gold-standard method of causal inference that controls for both unknown and unmeasured confounders—the randomized controlled trial—is infeasible for the key questions being asked. Other causal inference methods that rely on various natural experiments and have the major advantage of controlling for some or all unknown confounders—e.g., instrumental variable or co-relative analyses (
3–
5)—are potentially feasible but often are not available. An instrumental variable design involves the identification of an “instrument” that predicts risk factor exposure but has no direct effect on the disease outcome. A recent example uses month of birth as the instrument that has an impact on academic attainment (the younger children in each school class performing more poorly), which in turn predicts risk for drug abuse (
6). Co-relative designs control for familial confounders by comparing the risk for disease in an exposed case subject with that seen in an unexposed close relative, ideally, a sibling or monozygotic twin.
So, in the absence of more definitive methods to control for confounding, it behooves the critical reader to review both the background and the methodologic features of the study to help weigh the probability of a causal relationship between the risk factors and the outcome. Several relevant features here are noteworthy. First, this study is situated in a long line of investigations examining the impact of prenatal viral and bacterial infections on risk for psychosis in offspring (
2). While there are some differences across the many studies, the general consistency of findings provides support for causal processes. Second, several methodological features of this particular study population, including sampling, exclusion of cases of organic or drug-induced psychosis, and methods of follow-up and diagnosis, appear to have been carefully performed. Third, a body of basic science findings, briefly reviewed by Lee et al. and by Brown and Derkits (
2), provides support for the plausibility of the effects seen and postulates a potential disease mechanism. Fourth, the authors performed a sensitivity analysis for their assessment of bacterial infections, which supported the robustness of their measure of exposure. Fifth, they found a dose-response relationship, with systemic infections having a considerably stronger effect than localized infections. Such results have long been considered an important feature of causal risk factors, as far back as the classical debate about the role of smoking in lung cancer (
7).
What in this study might raise concerns about the causal effects of prenatal bacterial infections on psychosis risk? The greatest concern is the lack of replication across sexes. The sex differences seen here are striking and statistically significant. In the key analysis for any bacterial infection, the adjusted odds ratio in females is estimated at 1.0—that is, completely null. The confidence intervals are wide, and the authors appropriately caution us against overinterpretation. However, the common-sense conclusion is that the postulated effect occurs for male offspring but not for females. In many theories of causality, the generalizability of the causal claim is considered to be one important characteristic. A causal effect that applies across a wide range of situations is a deeper, more robust, and ultimately more useful explanation (
8).
The authors do provide a plausible account of the possible origins of this discrepancy and find similar trends in their other work on this cohort. However, the most recent extensive review of the literature on prenatal infections and risk for schizophrenia (
2) does not provide much precedent for the large sex effects detected in the Lee et al. study. Furthermore, we now know with considerable confidence that the genetic risk factors for schizophrenia are very highly correlated in men and women (
9). So this remains a somewhat troubling feature of their findings. A less important but noteworthy concern about their findings is their inability to replicate evidence from a number of previous studies that the impact of prenatal infections on schizophrenia is significantly greater in those at high familial risk. However, the study is not likely well powered to detect modest interaction effects, and, as mentioned above, the authors note that their measures of parental psychiatric illness are of limited quality.
In summary, this is a methodologically strong study that further advances our knowledge about what is likely an important environmental etiologic pathway to psychotic illness. Some skepticism is warranted because the authors’ causal inference depends entirely upon their ability to include in their multiple regression models all the relevant confounders and because of the large and somewhat puzzling difference in their results in males and females. The long line of prior work on this topic and the authors’ demonstration of a clear dose-response relationship between prenatal infection and offspring psychosis risk appropriately provide further evidence for the likely validity of their findings. The importance of their work is clear given the possibility of translating these findings—if further confirmed by larger and more definitive investigations—into primary prevention efforts.